

Small Molecule-mediated Insulin Hypersecretion Induces Transient ER Stress Response and Loss of Beta Cell Function
- PMID: 35641126
- PMCID: PMC9225822
- DOI: 10.1210/endocr/bqac081
Small Molecule-mediated Insulin Hypersecretion Induces Transient ER Stress Response and Loss of Beta Cell Function
Abstract
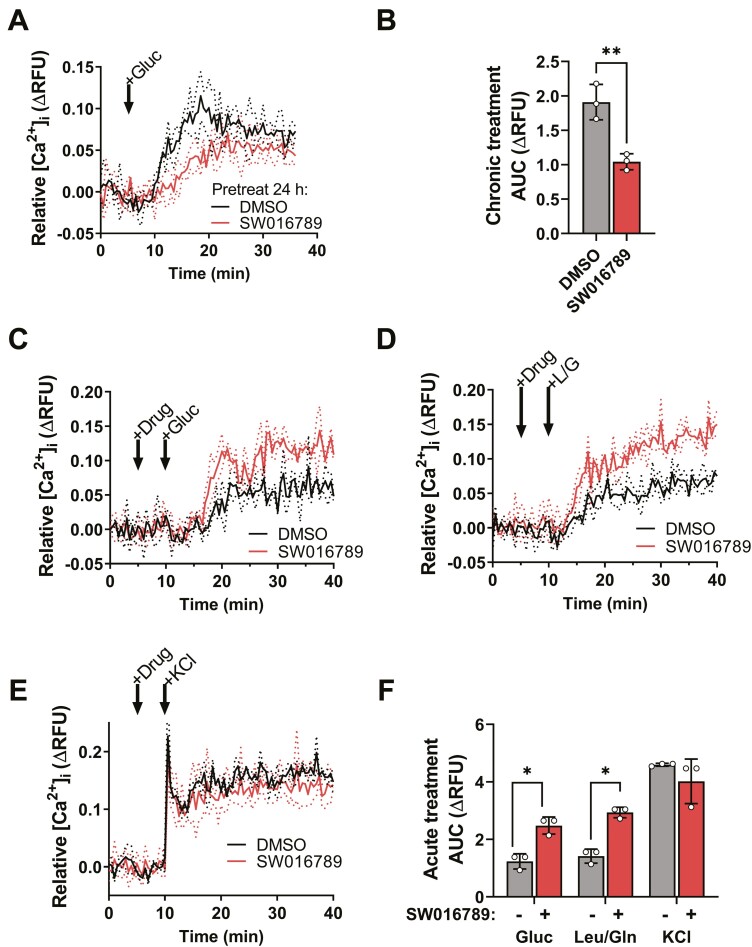
Pancreatic islet beta cells require a fine-tuned endoplasmic reticulum (ER) stress response for normal function; abnormal ER stress contributes to diabetes pathogenesis. Here, we identified a small molecule, SW016789, with time-dependent effects on beta cell ER stress and function. Acute treatment with SW016789 potentiated nutrient-induced calcium influx and insulin secretion, while chronic exposure to SW016789 transiently induced ER stress and shut down secretory function in a reversible manner. Distinct from the effects of thapsigargin, SW016789 did not affect beta cell viability or apoptosis, potentially due to a rapid induction of adaptive genes, weak signaling through the eIF2α kinase PERK, and lack of oxidative stress gene Txnip induction. We determined that SW016789 acted upstream of voltage-dependent calcium channels (VDCCs) and potentiated nutrient- but not KCl-stimulated calcium influx. Measurements of metabolomics, oxygen consumption rate, and G protein-coupled receptor signaling did not explain the potentiating effects of SW016789. In chemical cotreatment experiments, we discovered synergy between SW016789 and activators of protein kinase C and VDCCs, suggesting involvement of these pathways in the mechanism of action. Finally, chronically elevated calcium influx was required for the inhibitory impact of SW016789, as blockade of VDCCs protected human islets and MIN6 beta cells from hypersecretion-induced dysfunction. We conclude that beta cells undergoing this type of pharmacological hypersecretion have the capacity to suppress their function to mitigate ER stress and avoid apoptosis. These results have the potential to uncover beta cell ER stress mitigation factors and add support to beta cell rest strategies to preserve function.
Keywords: endoplasmic reticulum stress; insulin secretion; pancreatic islet beta cells.
© The Author(s) 2022. Published by Oxford University Press on behalf of the Endocrine Society. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
Figures







References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
