

Nuclear antiviral innate responses at the intersection of DNA sensing and DNA repair
- PMID: 35641341
- PMCID: PMC9560981
- DOI: 10.1016/j.tim.2022.05.004
Nuclear antiviral innate responses at the intersection of DNA sensing and DNA repair
Abstract
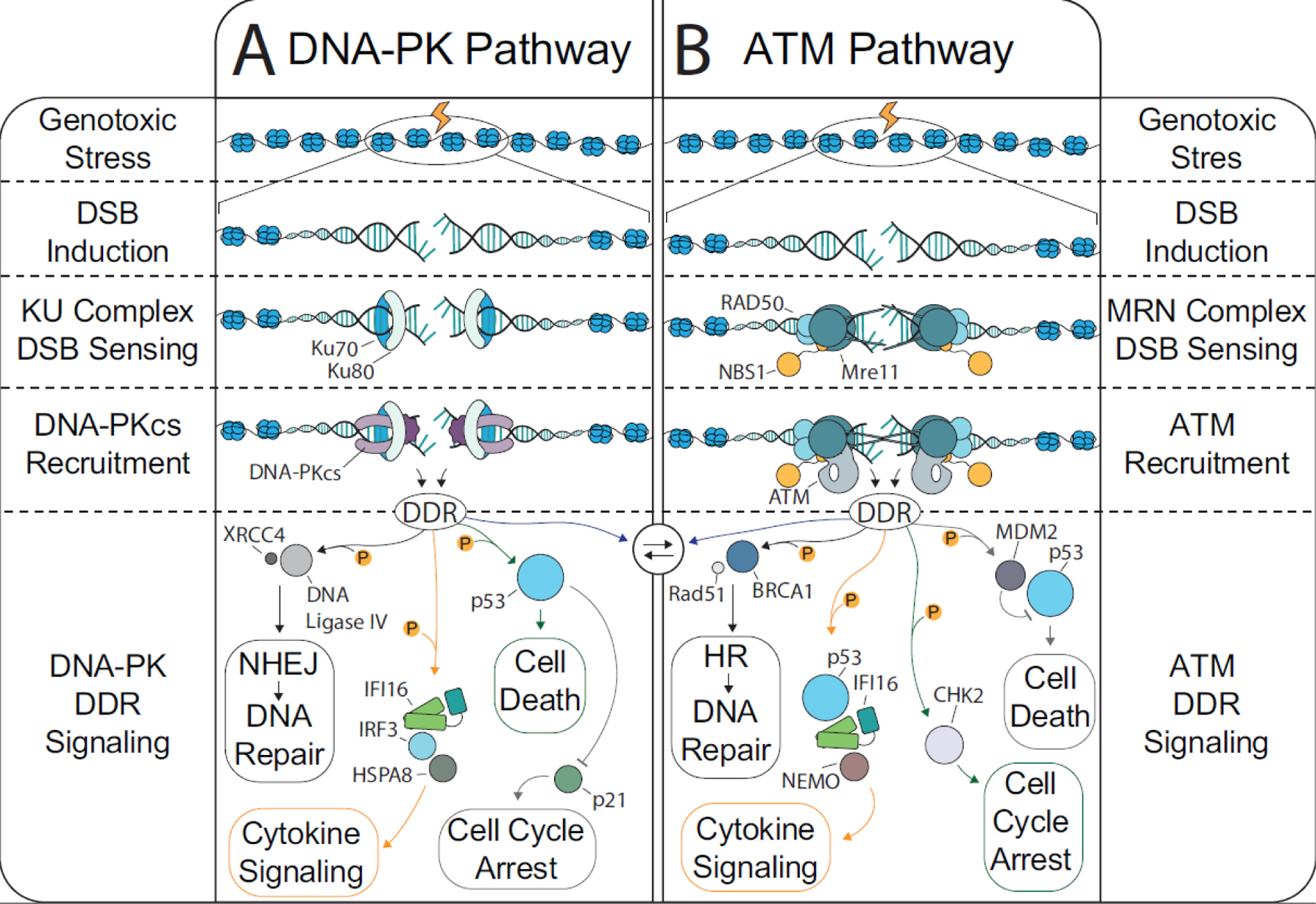
The coevolution of vertebrate and mammalian hosts with DNA viruses has driven the ability of host cells to distinguish viral from cellular DNA in the nucleus to induce intrinsic immune responses. Concomitant viral mechanisms have arisen to inhibit DNA sensing. At this virus-host interface, emerging evidence links cytokine responses and cellular homeostasis pathways, particularly the DNA damage response (DDR). Nuclear DNA sensors, such as the interferon (IFN)-γ inducible protein 16 (IFI16), functionally intersect with the DDR regulators ataxia telangiectasia mutated (ATM) and DNA-dependent protein kinase (DNA-PK). Here, we discuss accumulating knowledge for the DDR-innate immunity signaling axis. Through the lens of this infection-driven signaling axis, we present host and viral molecular strategies acquired to regulate autoinflammation and antiviral responses.
Keywords: DNA damage response; DNA virus infection; antiviral response; interferon; nuclear DNA sensing.
Copyright © 2022 Elsevier Ltd. All rights reserved.
Conflict of interest statement
Declaration of interests No interests are declared.
Figures



References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
