

Structural variant-based pangenome construction has low sensitivity to variability of haplotype-resolved bovine assemblies
- PMID: 35641504
- PMCID: PMC9156671
- DOI: 10.1038/s41467-022-30680-2
Structural variant-based pangenome construction has low sensitivity to variability of haplotype-resolved bovine assemblies
Abstract
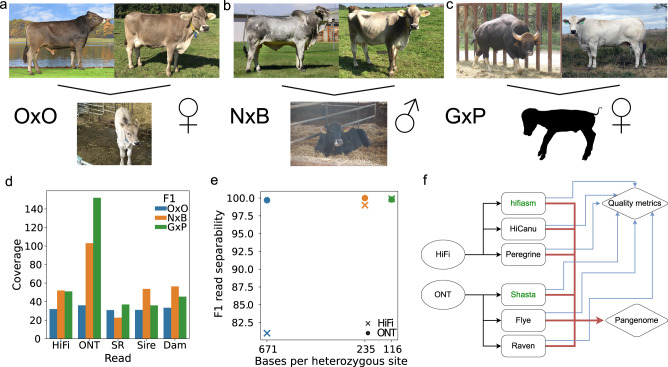
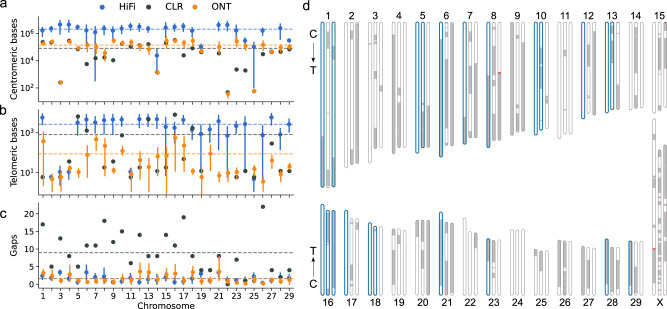
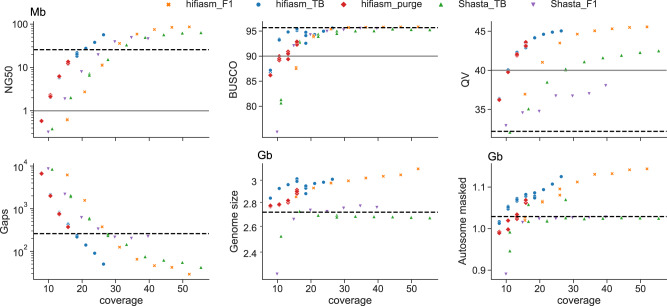
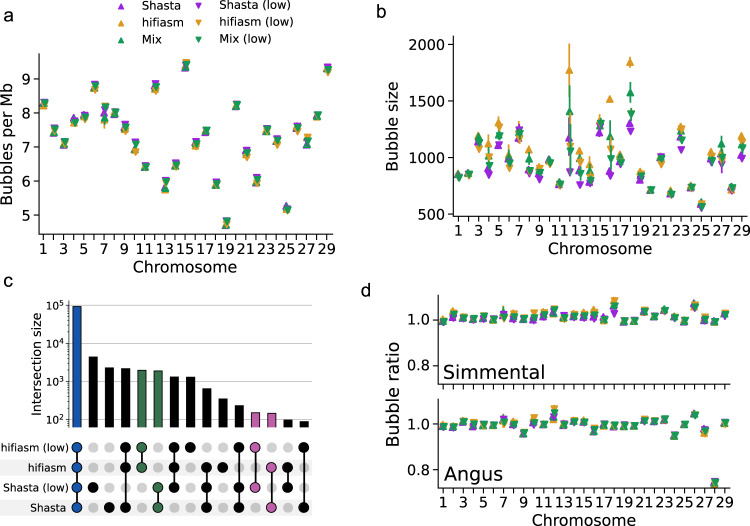
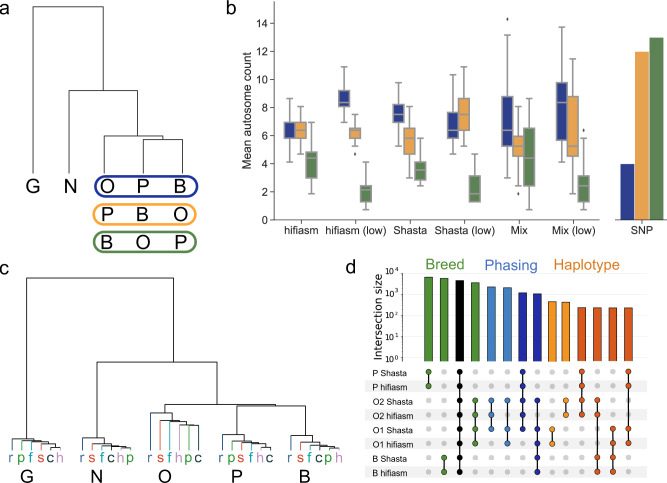
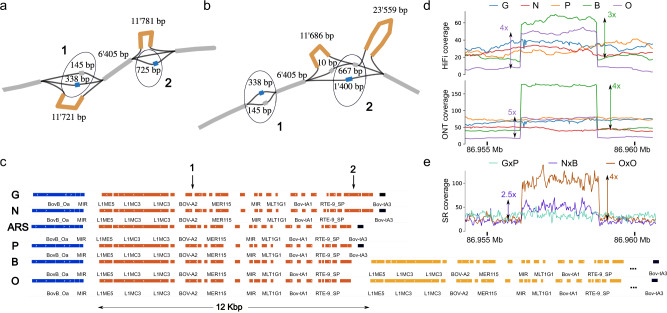
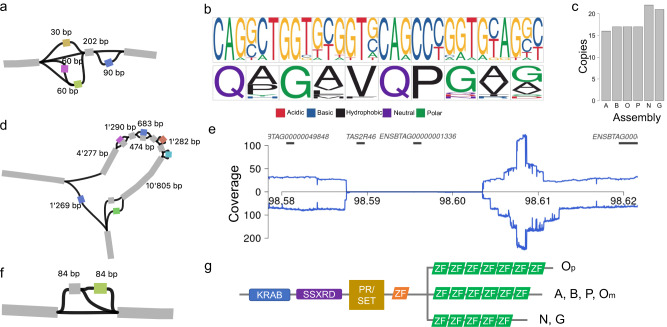
Advantages of pangenomes over linear reference assemblies for genome research have recently been established. However, potential effects of sequence platform and assembly approach, or of combining assemblies created by different approaches, on pangenome construction have not been investigated. Here we generate haplotype-resolved assemblies from the offspring of three bovine trios representing increasing levels of heterozygosity that each demonstrate a substantial improvement in contiguity, completeness, and accuracy over the current Bos taurus reference genome. Diploid coverage as low as 20x for HiFi or 60x for ONT is sufficient to produce two haplotype-resolved assemblies meeting standards set by the Vertebrate Genomes Project. Structural variant-based pangenomes created from the haplotype-resolved assemblies demonstrate significant consensus regardless of sequence platform, assembler algorithm, or coverage. Inspecting pangenome topologies identifies 90 thousand structural variants including 931 overlapping with coding sequences; this approach reveals variants affecting QRICH2, PRDM9, HSPA1A, TAS2R46, and GC that have potential to affect phenotype.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
