

Genomic dissection of Klebsiella pneumoniae infections in hospital patients reveals insights into an opportunistic pathogen
- PMID: 35641522
- PMCID: PMC9156735
- DOI: 10.1038/s41467-022-30717-6
Genomic dissection of Klebsiella pneumoniae infections in hospital patients reveals insights into an opportunistic pathogen
Abstract
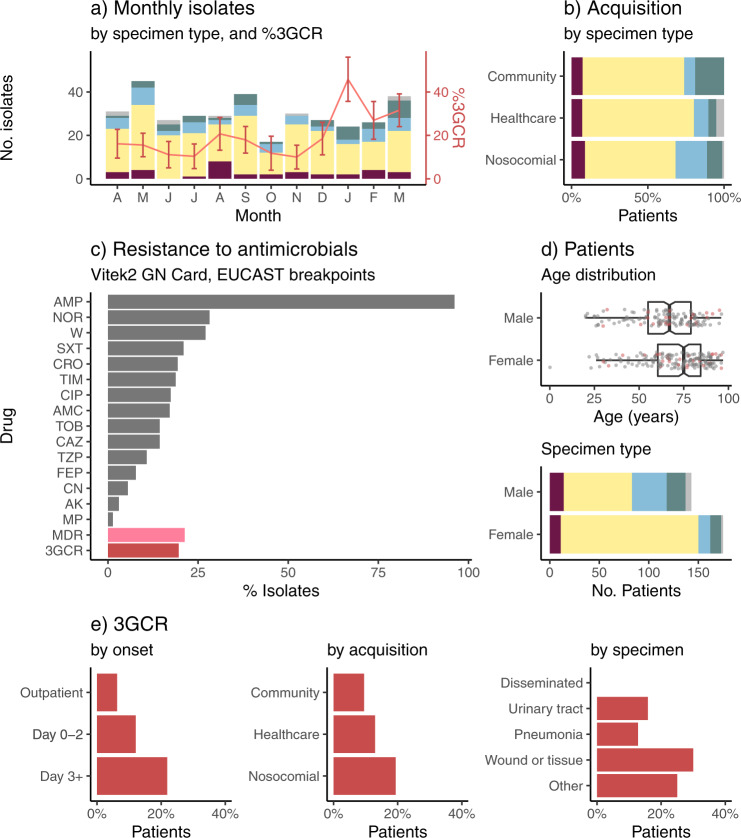
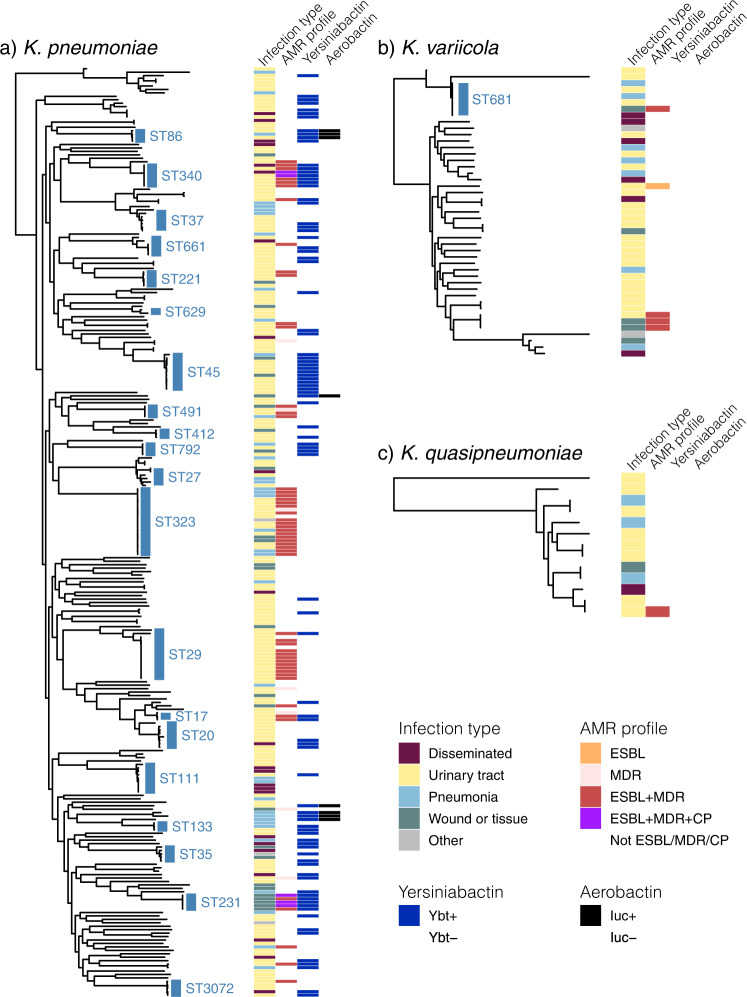
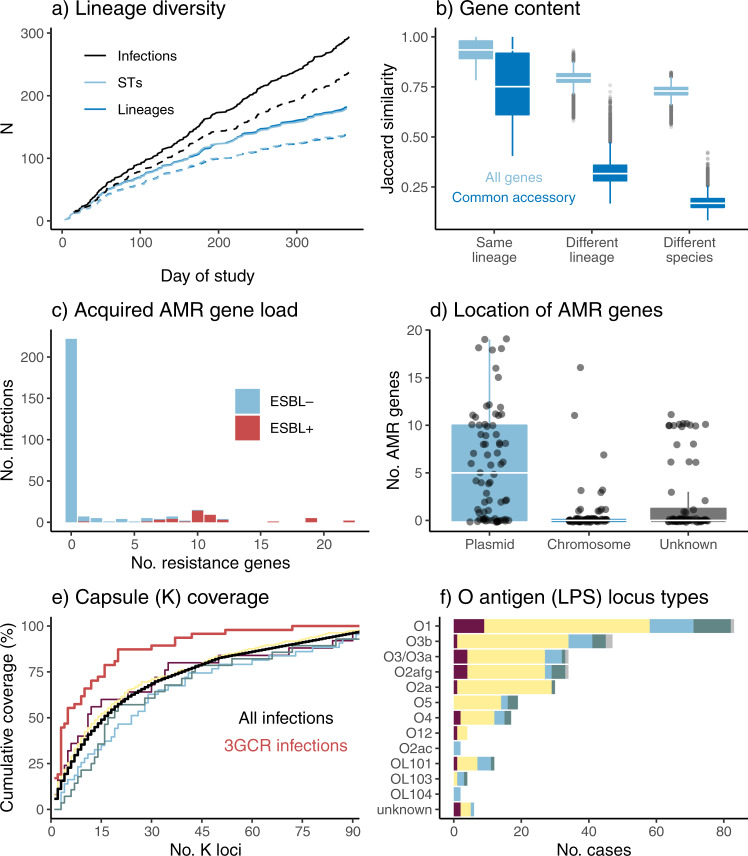
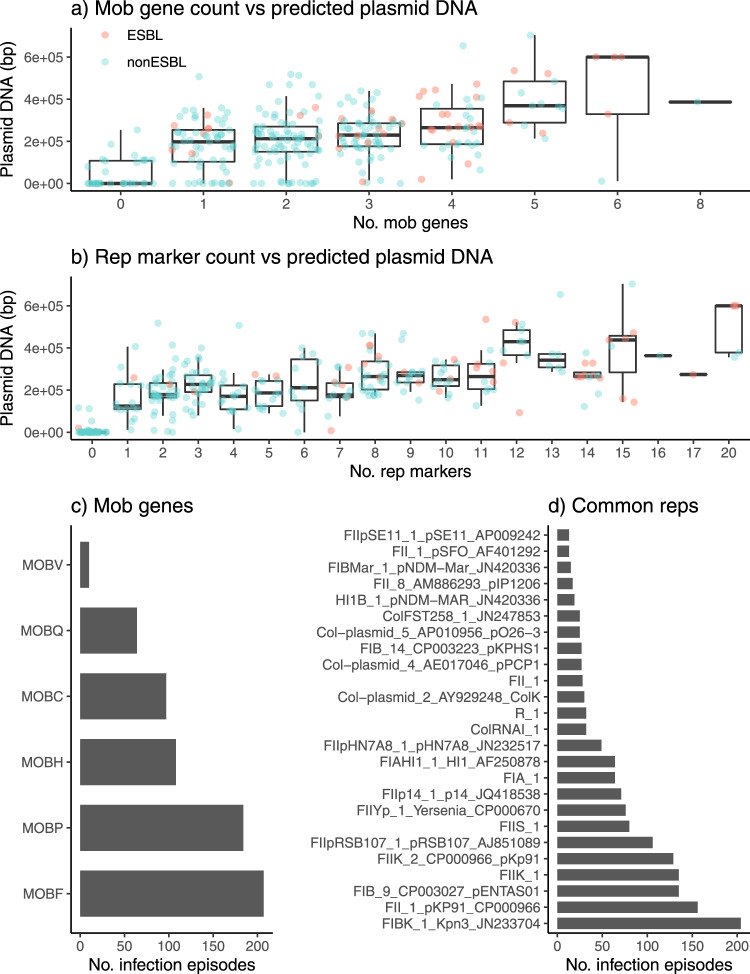
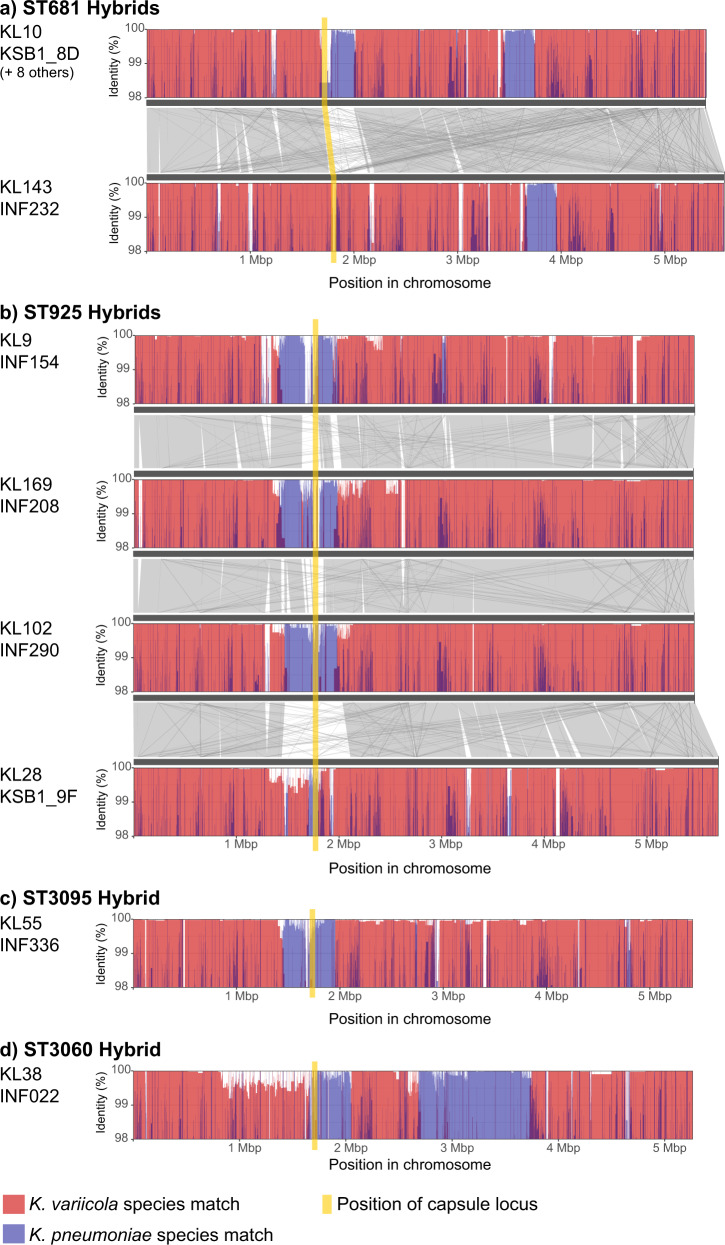
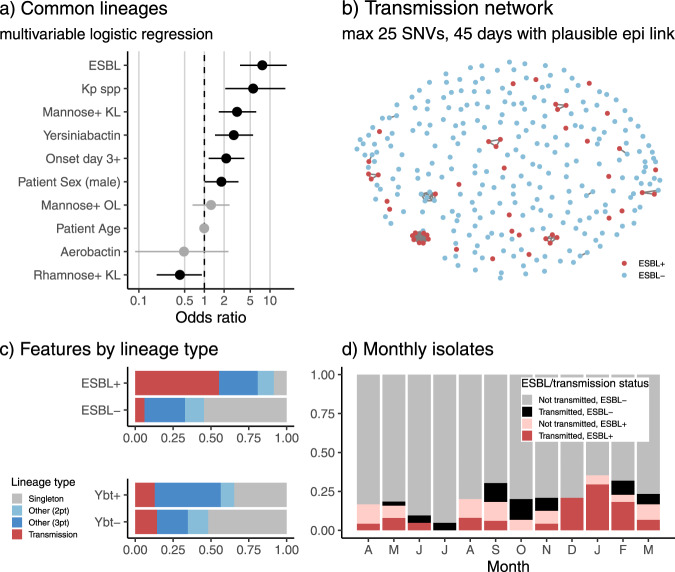
Klebsiella pneumoniae is a major cause of opportunistic healthcare-associated infections, which are increasingly complicated by the presence of extended-spectrum beta-lactamases (ESBLs) and carbapenem resistance. We conducted a year-long prospective surveillance study of K. pneumoniae clinical isolates in hospital patients. Whole-genome sequence (WGS) data reveals a diverse pathogen population, including other species within the K. pneumoniae species complex (18%). Several infections were caused by K. variicola/K. pneumoniae hybrids, one of which shows evidence of nosocomial transmission. A wide range of antimicrobial resistance (AMR) phenotypes are observed, and diverse genetic mechanisms identified (mainly plasmid-borne genes). ESBLs are correlated with presence of other acquired AMR genes (median n = 10). Bacterial genomic features associated with nosocomial onset are ESBLs (OR 2.34, p = 0.015) and rhamnose-positive capsules (OR 3.12, p < 0.001). Virulence plasmid-encoded features (aerobactin, hypermucoidy) are observed at low-prevalence (<3%), mostly in community-onset cases. WGS-confirmed nosocomial transmission is implicated in just 10% of cases, but strongly associated with ESBLs (OR 21, p < 1 × 10-11). We estimate 28% risk of onward nosocomial transmission for ESBL-positive strains vs 1.7% for ESBL-negative strains. These data indicate that K. pneumoniae infections in hospitalised patients are due largely to opportunistic infections with diverse strains, with an additional burden from nosocomially-transmitted AMR strains and community-acquired hypervirulent strains.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
