

High-resolution structures of the SAMHD1 dGTPase homolog from Leeuwenhoekiella blandensis reveal a novel mechanism of allosteric activation by dATP
- PMID: 35643313
- PMCID: PMC9257424
- DOI: 10.1016/j.jbc.2022.102073
High-resolution structures of the SAMHD1 dGTPase homolog from Leeuwenhoekiella blandensis reveal a novel mechanism of allosteric activation by dATP
Abstract
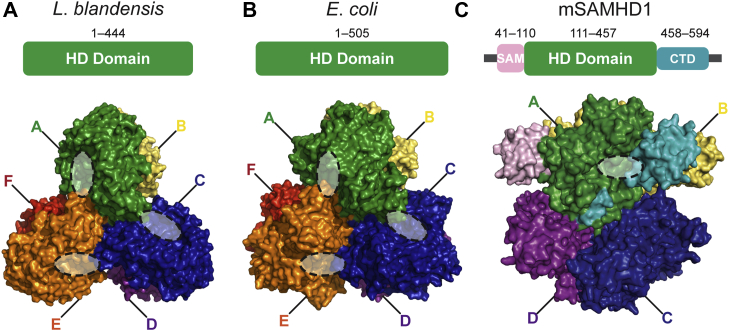
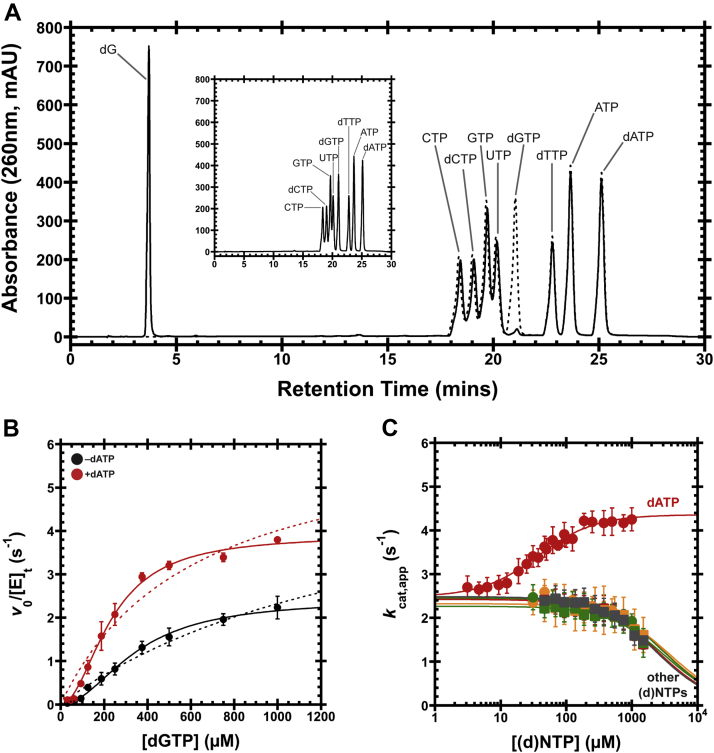
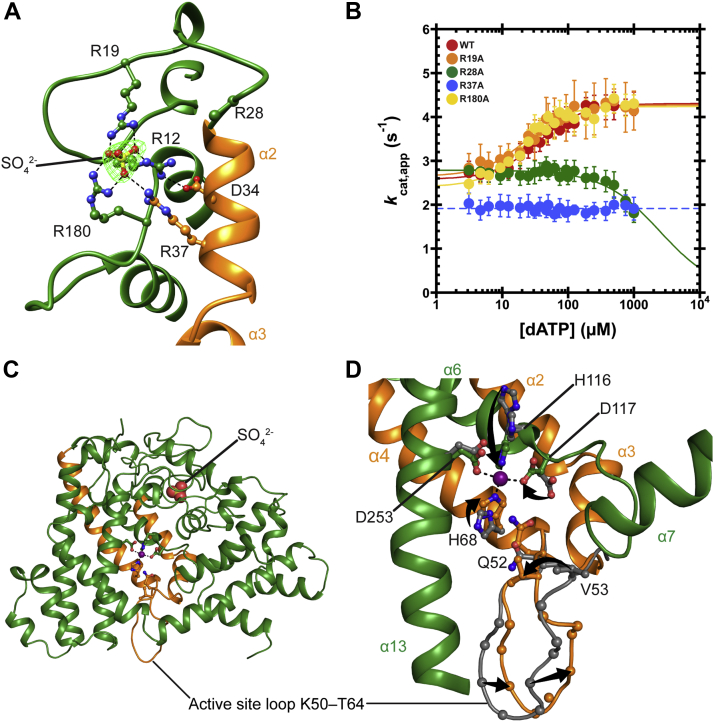
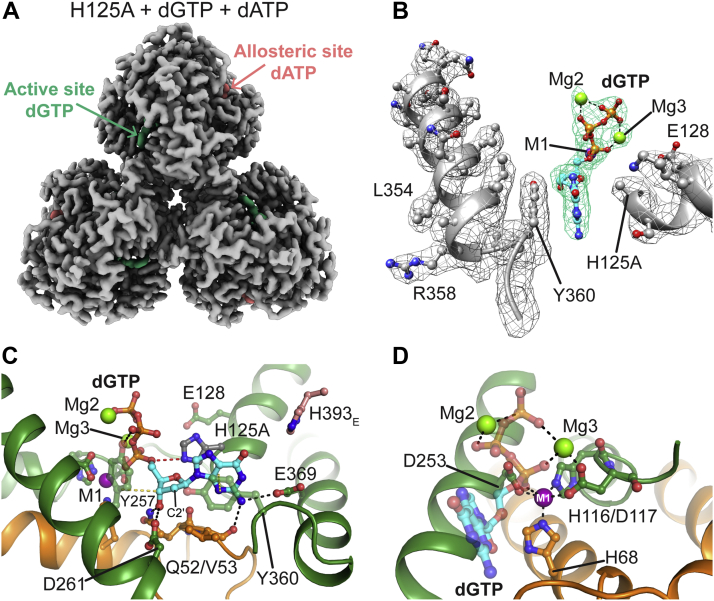
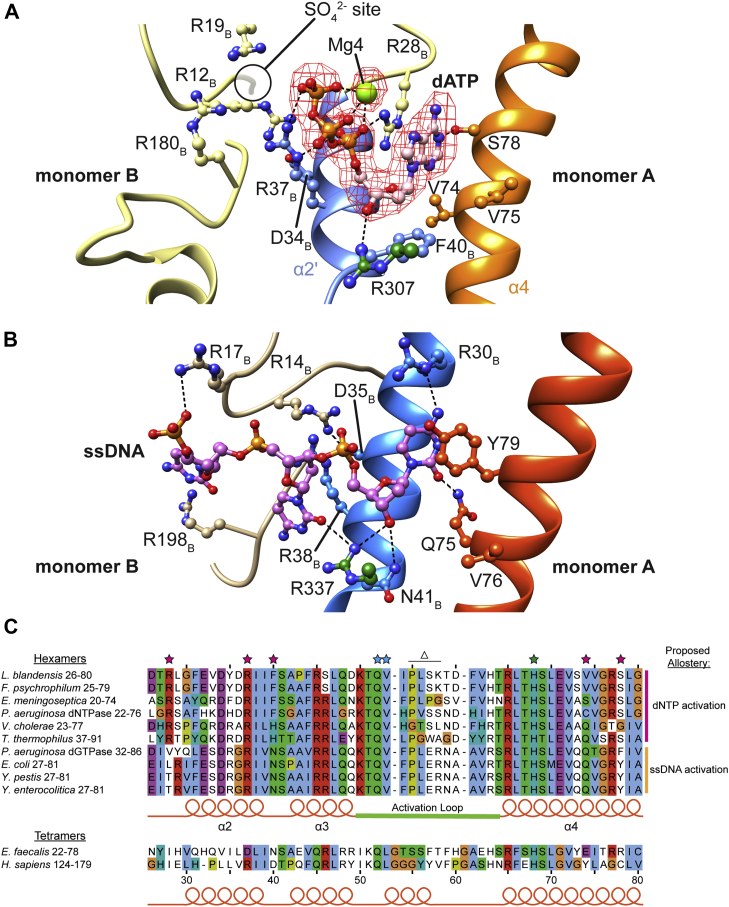
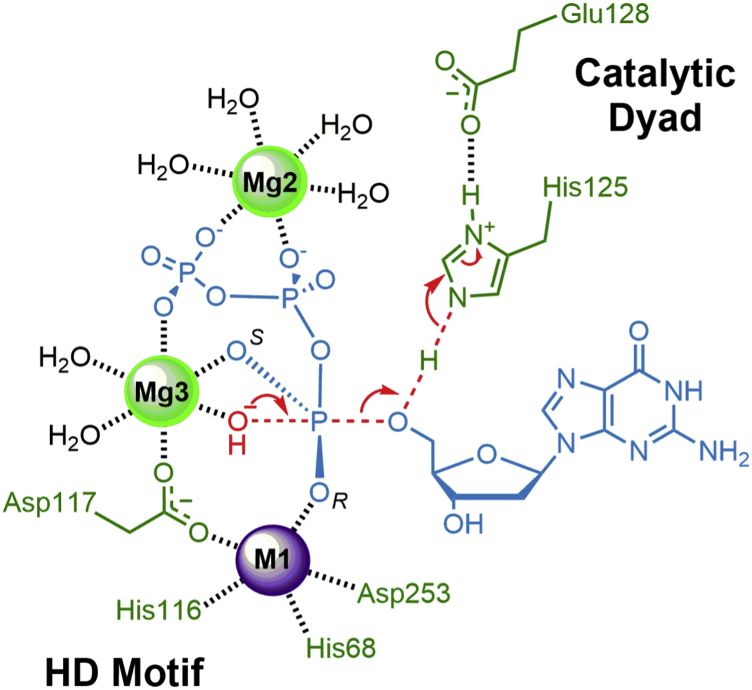
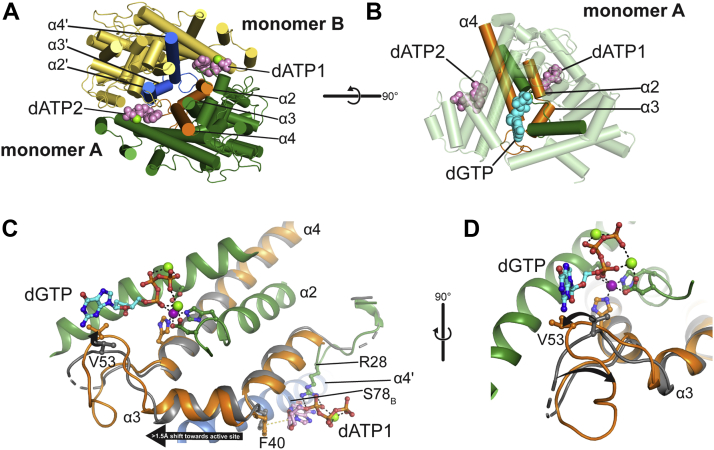
Deoxynucleoside triphosphate (dNTP) triphosphohydrolases (dNTPases) are important enzymes that may perform multiple functions in the cell, including regulating the dNTP pools and contributing to innate immunity against viruses. Among the homologs that are best studied are human sterile alpha motif and HD domain-containing protein 1 (SAMHD1), a tetrameric dNTPase, and the hexameric Escherichia coli dGTPase; however, it is unclear whether these are representative of all dNTPases given their wide distribution throughout life. Here, we investigated a hexameric homolog from the marine bacterium Leeuwenhoekiella blandensis, revealing that it is a dGTPase that is subject to allosteric activation by dATP, specifically. Allosteric regulation mediated solely by dATP represents a novel regulatory feature among dNTPases that may facilitate maintenance of cellular dNTP pools in L. blandensis. We present high-resolution X-ray crystallographic structures (1.80-2.26 Å) in catalytically important conformations as well as cryo-EM structures (2.1-2.7 Å) of the enzyme bound to dGTP and dATP ligands. The structures, the highest resolution cryo-EM structures of any SAMHD1-like dNTPase to date, reveal an intact metal-binding site with the dGTP substrate coordinated to three metal ions. These structural and biochemical data yield insights into the catalytic mechanism and support a conserved catalytic mechanism for the tetrameric and hexameric dNTPase homologs. We conclude that the allosteric activation by dATP appears to rely on structural connectivity between the allosteric and active sites, as opposed to the changes in oligomeric state upon ligand binding used by SAMHD1.
Keywords: allosteric regulation; cryo-EM; crystallography; enzyme mechanism; enzyme structure; nucleoside/nucleotide metabolism; structure–function.
Published by Elsevier Inc.
Conflict of interest statement
Conflict of interest The authors declare that they have no conflicts of interest with the contents of this article.
Figures
Similar articles
-
The crystal structure of dGTPase reveals the molecular basis of dGTP selectivity.Proc Natl Acad Sci U S A. 2019 May 7;116(19):9333-9339. doi: 10.1073/pnas.1814999116. Epub 2019 Apr 24. Proc Natl Acad Sci U S A. 2019. PMID: 31019074 Free PMC article.
-
Structural basis of allosteric activation of sterile α motif and histidine-aspartate domain-containing protein 1 (SAMHD1) by nucleoside triphosphates.J Biol Chem. 2014 Nov 21;289(47):32617-27. doi: 10.1074/jbc.M114.591958. Epub 2014 Oct 6. J Biol Chem. 2014. PMID: 25288794 Free PMC article.
-
Structural basis of cellular dNTP regulation by SAMHD1.Proc Natl Acad Sci U S A. 2014 Oct 14;111(41):E4305-14. doi: 10.1073/pnas.1412289111. Epub 2014 Sep 29. Proc Natl Acad Sci U S A. 2014. PMID: 25267621 Free PMC article.
-
The missing link: allostery and catalysis in the anti-viral protein SAMHD1.Biochem Soc Trans. 2019 Aug 30;47(4):1013-1027. doi: 10.1042/BST20180348. Epub 2019 Jul 11. Biochem Soc Trans. 2019. PMID: 31296733 Free PMC article. Review.
-
SAMHD1 in cancer: curse or cure?J Mol Med (Berl). 2022 Mar;100(3):351-372. doi: 10.1007/s00109-021-02131-w. Epub 2021 Sep 4. J Mol Med (Berl). 2022. PMID: 34480199 Free PMC article. Review.
Cited by
-
Guanine-containing ssDNA and RNA induce dimeric and tetrameric structural forms of SAMHD1.Nucleic Acids Res. 2023 Dec 11;51(22):12443-12458. doi: 10.1093/nar/gkad971. Nucleic Acids Res. 2023. PMID: 37930833 Free PMC article.
-
Reaction Mechanism and Metal Selectivity of Human SAMHD1 Elucidated by QM/MM Calculations.ACS Catal. 2025 Jun 1;15(12):10176-10187. doi: 10.1021/acscatal.5c01682. eCollection 2025 Jun 20. ACS Catal. 2025. PMID: 40568220 Free PMC article.
-
A deoxynucleoside triphosphate triphosphohydrolase promotes cell cycle progression in Caulobacter crescentus.J Bacteriol. 2025 Jun 24;207(6):e0014525. doi: 10.1128/jb.00145-25. Epub 2025 Jun 2. J Bacteriol. 2025. PMID: 40454833 Free PMC article.
-
A deoxynucleoside triphosphate triphosphohydrolase promotes cell cycle progression in Caulobacter crescentus.bioRxiv [Preprint]. 2024 Apr 26:2024.04.25.591158. doi: 10.1101/2024.04.25.591158. bioRxiv. 2024. Update in: J Bacteriol. 2025 Jun 24;207(6):e0014525. doi: 10.1128/jb.00145-25. PMID: 38712277 Free PMC article. Updated. Preprint.
-
Mechanism by which T7 bacteriophage protein Gp1.2 inhibits Escherichia coli dGTPase.Proc Natl Acad Sci U S A. 2022 Sep 13;119(37):e2123092119. doi: 10.1073/pnas.2123092119. Epub 2022 Sep 6. Proc Natl Acad Sci U S A. 2022. PMID: 36067314 Free PMC article.
References
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
