

Imbalanced prostanoid release mediates cigarette smoke-induced human pulmonary artery cell proliferation
- PMID: 35643499
- PMCID: PMC9145181
- DOI: 10.1186/s12931-022-02056-z
Imbalanced prostanoid release mediates cigarette smoke-induced human pulmonary artery cell proliferation
Abstract
Background: Pulmonary hypertension is a common and serious complication of chronic obstructive pulmonary disease (COPD). Studies suggest that cigarette smoke can initiate pulmonary vascular remodelling by stimulating cell proliferation; however, the underlying cause, particularly the role of vasoactive prostanoids, is unclear. We hypothesize that cigarette smoke extract (CSE) can induce imbalanced vasoactive prostanoid release by differentially modulating the expression of respective synthase genes in human pulmonary artery smooth muscle cells (PASMCs) and endothelial cells (PAECs), thereby contributing to cell proliferation.
Methods: Aqueous CSE was prepared from 3R4F research-grade cigarettes. Human PASMCs and PAECs were treated with or without CSE. Quantitative real-time RT-PCR and Western blotting were used to analyse the mRNA and protein expression of vasoactive prostanoid syhthases. Prostanoid concentration in the medium was measured using ELISA kits. Cell proliferation was assessed using the cell proliferation reagent WST-1.
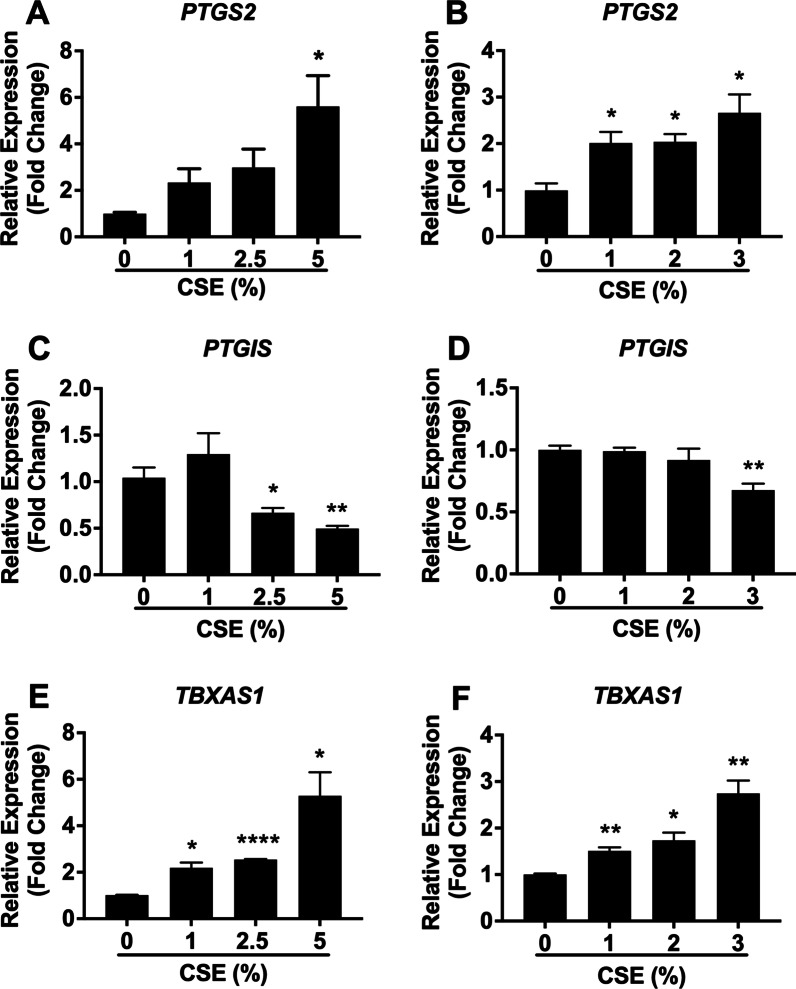
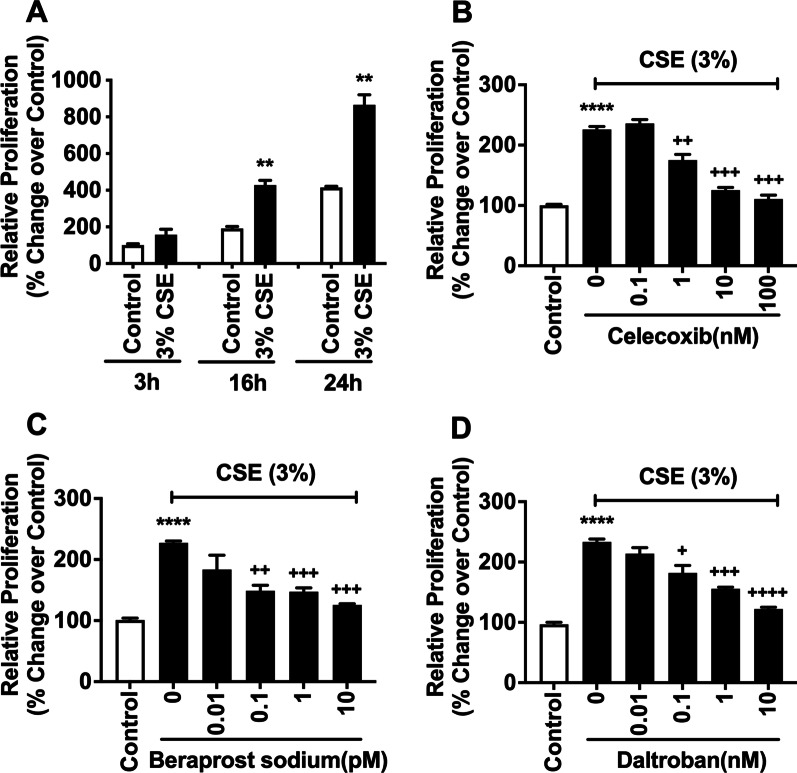
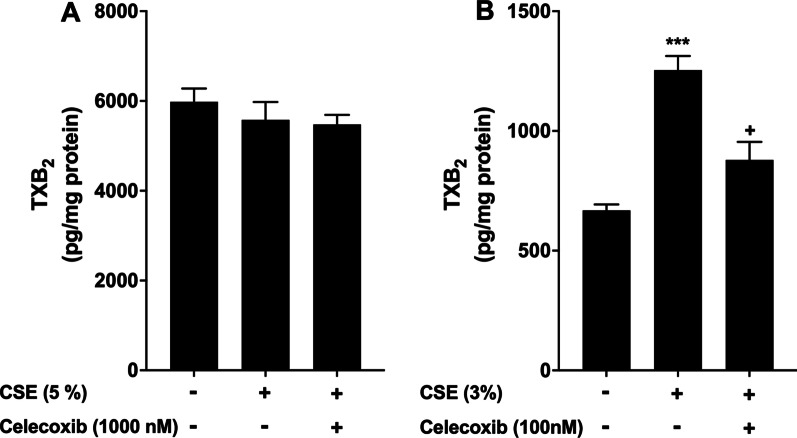
Results: We demonstrated that CSE induced the expression of cyclooxygenase-2 (COX-2), the rate-limiting enzyme in prostanoid synthesis, in both cell types. In PASMCs, CSE reduced the downstream prostaglandin (PG) I synthase (PGIS) mRNA and protein expression and PGI2 production, whereas in PAECs, CSE downregulated PGIS mRNA expression, but PGIS protein was undetectable and CSE had no effect on PGI2 production. CSE increased thromboxane (TX) A synthase (TXAS) mRNA expression and TXA2 production, despite undetectable TXAS protein in both cell types. CSE also reduced microsomal PGE synthase-1 (mPGES-1) protein expression and PGE2 production in PASMCs, but increased PGE2 production despite unchanged mPGES-1 protein expression in PAECs. Furthermore, CSE stimulated proliferation of both cell types, which was significantly inhibited by the selective COX-2 inhibitor celecoxib, the PGI2 analogue beraprost and the TXA2 receptor antagonist daltroban.
Conclusions: These findings provide the first evidence that cigarette smoke can induce imbalanced prostanoid mediator release characterized by the reduced PGI2/TXA2 ratio and contribute to pulmonary vascular remodelling and suggest that TXA2 may represent a novel therapeutic target for pulmonary hypertension in COPD.
Keywords: COPD; Cigarette smoke; Prostanoids; Pulmonary artery cell proliferation; Pulmonary hypertension.
© 2022. The Author(s).
Conflict of interest statement
All authors declare that they have no competing interests.
Figures




References
-
- Galie N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, et al. 2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: the Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT) Eur Heart J. 2016;37:67–119. doi: 10.1093/eurheartj/ehv317. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
