

Accounting for population structure in genetic studies of cystic fibrosis
- PMID: 35647563
- PMCID: PMC9136666
- DOI: 10.1016/j.xhgg.2022.100117
Accounting for population structure in genetic studies of cystic fibrosis
Abstract
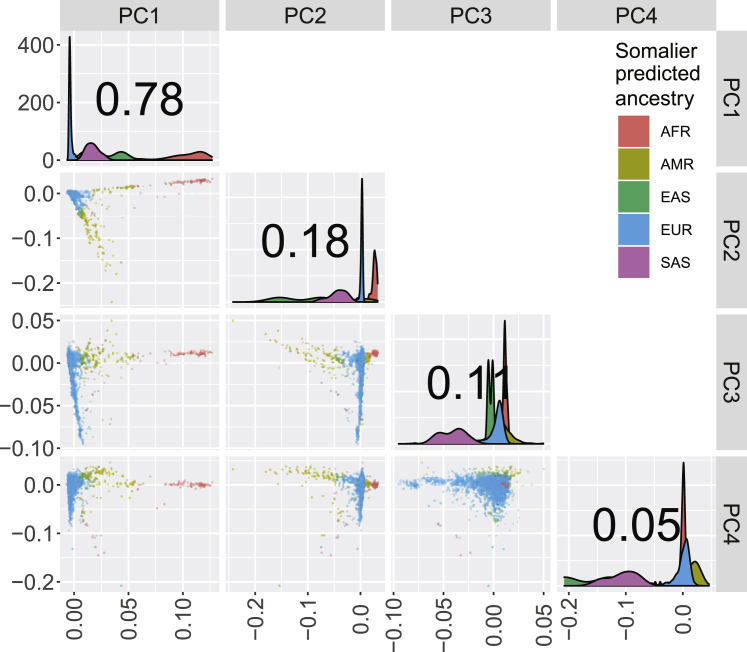
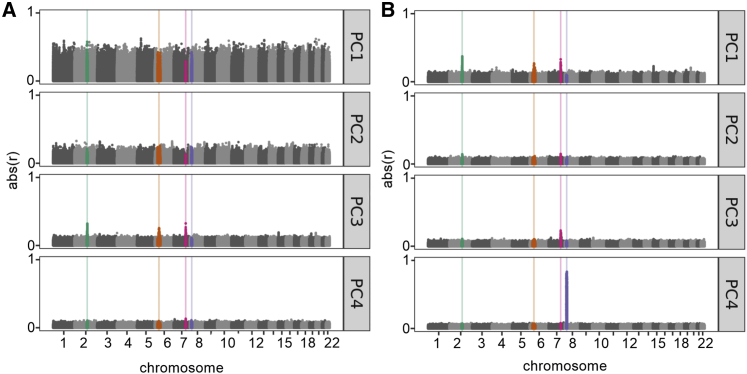
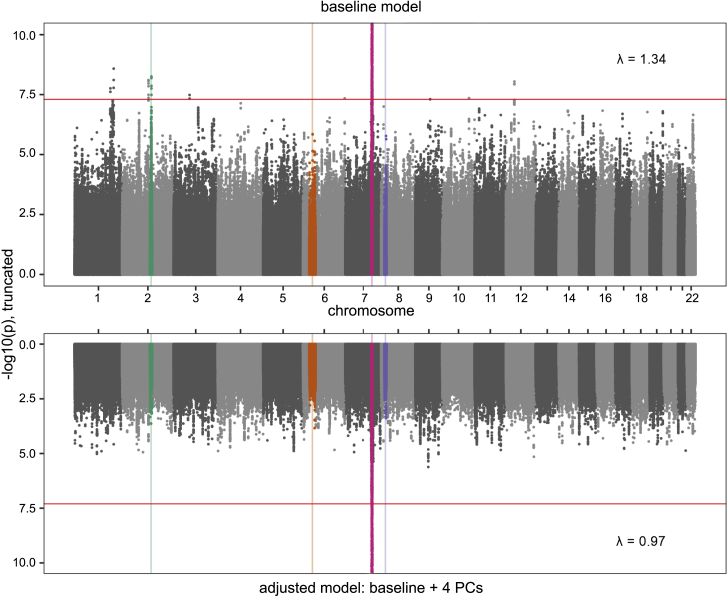
CFTR F508del (c.1521_1523delCTT, p.Phe508delPhe) is the most common pathogenic allele underlying cystic fibrosis (CF), and its frequency varies in a geographic cline across Europe. We hypothesized that genetic variation associated with this cline is overrepresented in a large cohort (N > 5,000) of persons with CF who underwent whole-genome sequencing and that this pattern could result in spurious associations between variants correlated with both the F508del genotype and CF-related outcomes. Using principal-component (PC) analyses, we showed that variation in the CFTR region disproportionately contributes to a PC explaining a relatively high proportion of genetic variance. Variation near CFTR was correlated with population structure among persons with CF, and this correlation was driven by a subset of the sample inferred to have European ancestry. We performed genome-wide association studies comparing persons with CF with one versus two copies of the F508del allele; this allowed us to identify genetic variation associated with the F508del allele and to determine that standard PC-adjustment strategies eliminated the significant association signals. Our results suggest that PC adjustment can adequately prevent spurious associations between genetic variants and CF-related traits and are therefore effective tools to control for population structure even when population structure is confounded with disease severity and a common pathogenic variant.
Keywords: CFTR F508del; genome-wide association study; population structure.
© 2022 The Author(s).
Conflict of interest statement
M.B. is the editor-in-chief and J.X.C. (member of the Cystic Fibrosis Genome Project) is the deputy editor of HGG Advances. The authors declare no other competing interests.
Figures
References
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
