

Divergent Viruses Discovered in Swine Alter the Understanding of Evolutionary History and Genetic Diversity of the Respirovirus Genus and Related Porcine Parainfluenza Viruses
- PMID: 35647875
- PMCID: PMC9241844
- DOI: 10.1128/spectrum.00242-22
Divergent Viruses Discovered in Swine Alter the Understanding of Evolutionary History and Genetic Diversity of the Respirovirus Genus and Related Porcine Parainfluenza Viruses
Abstract
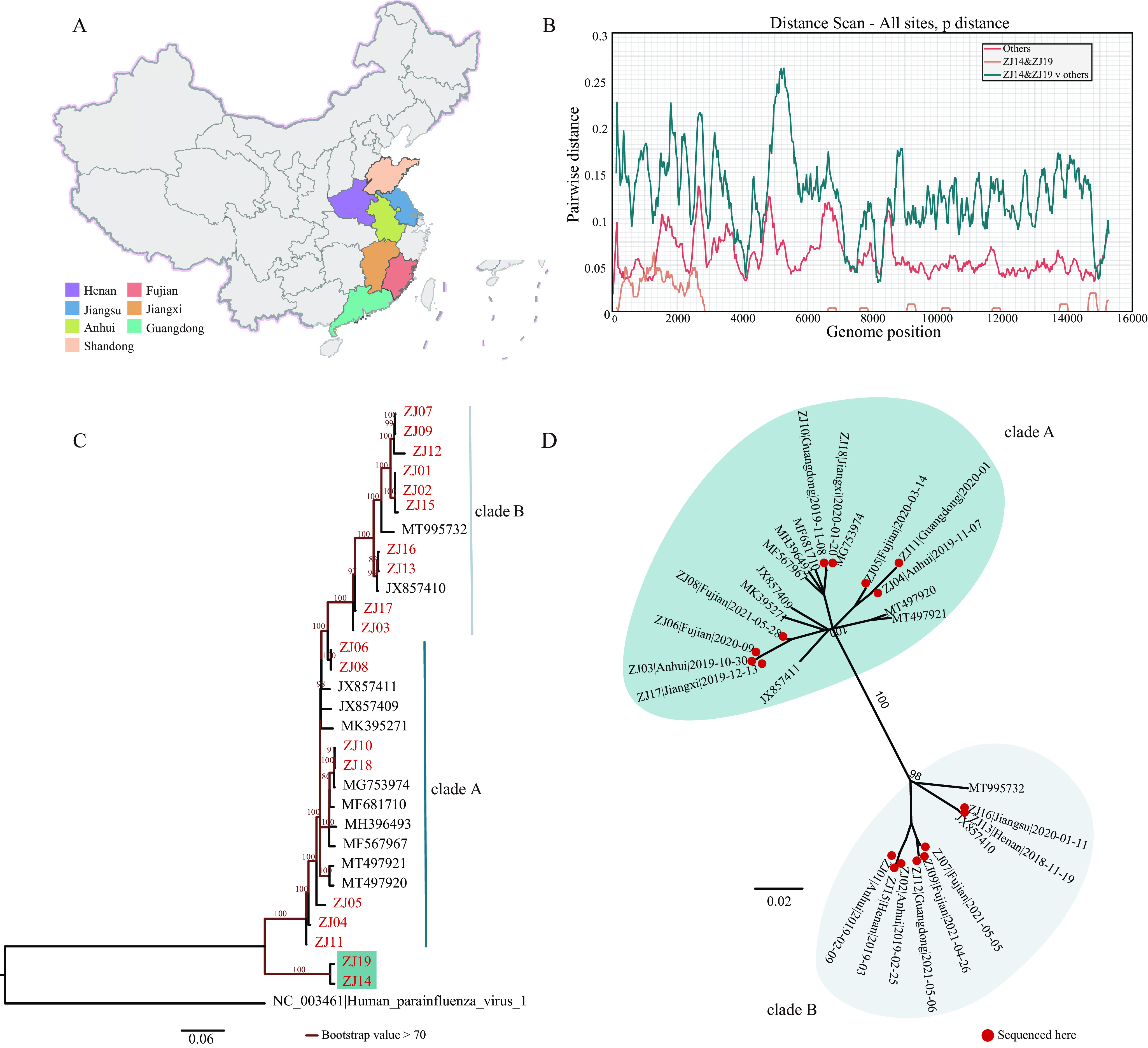
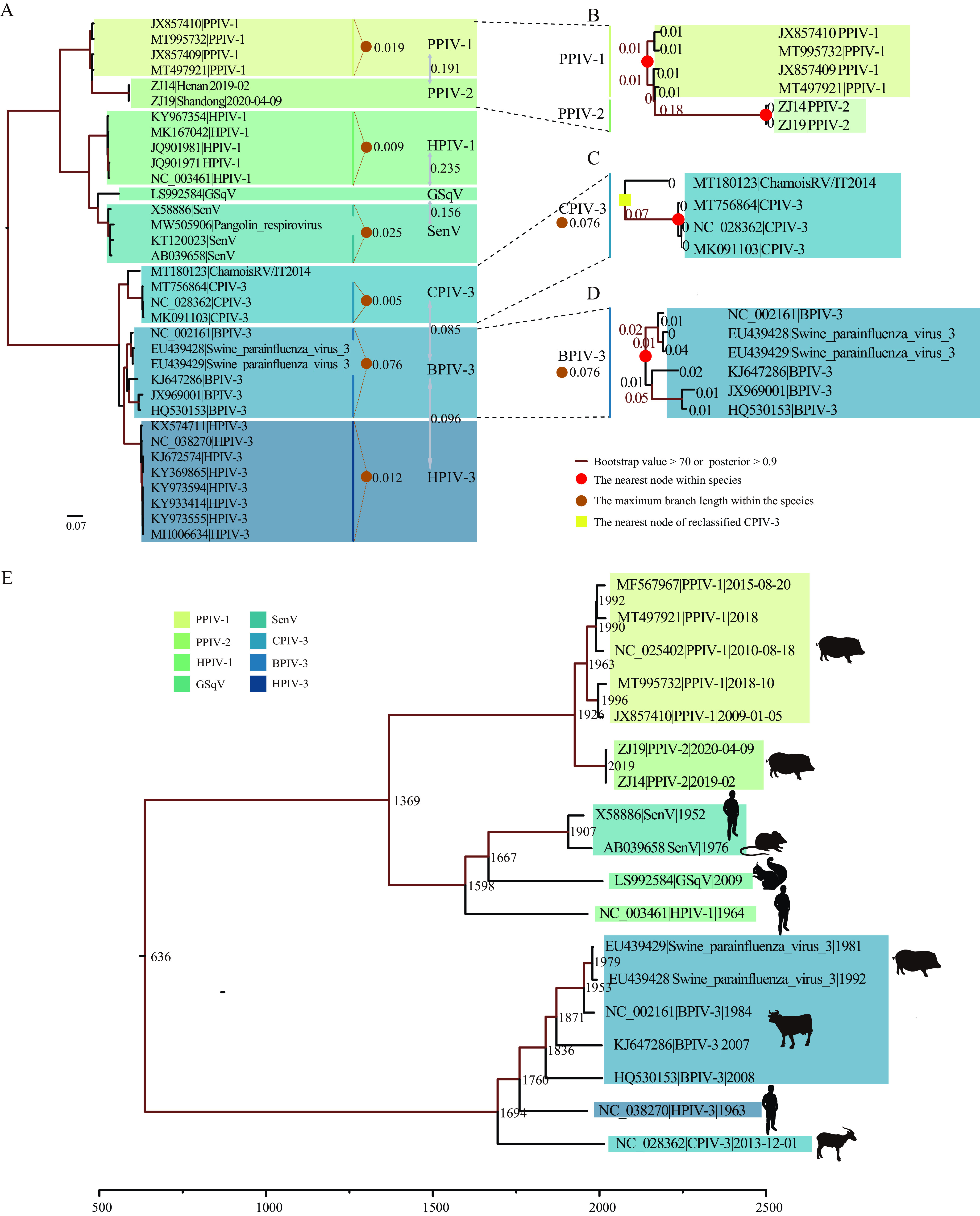
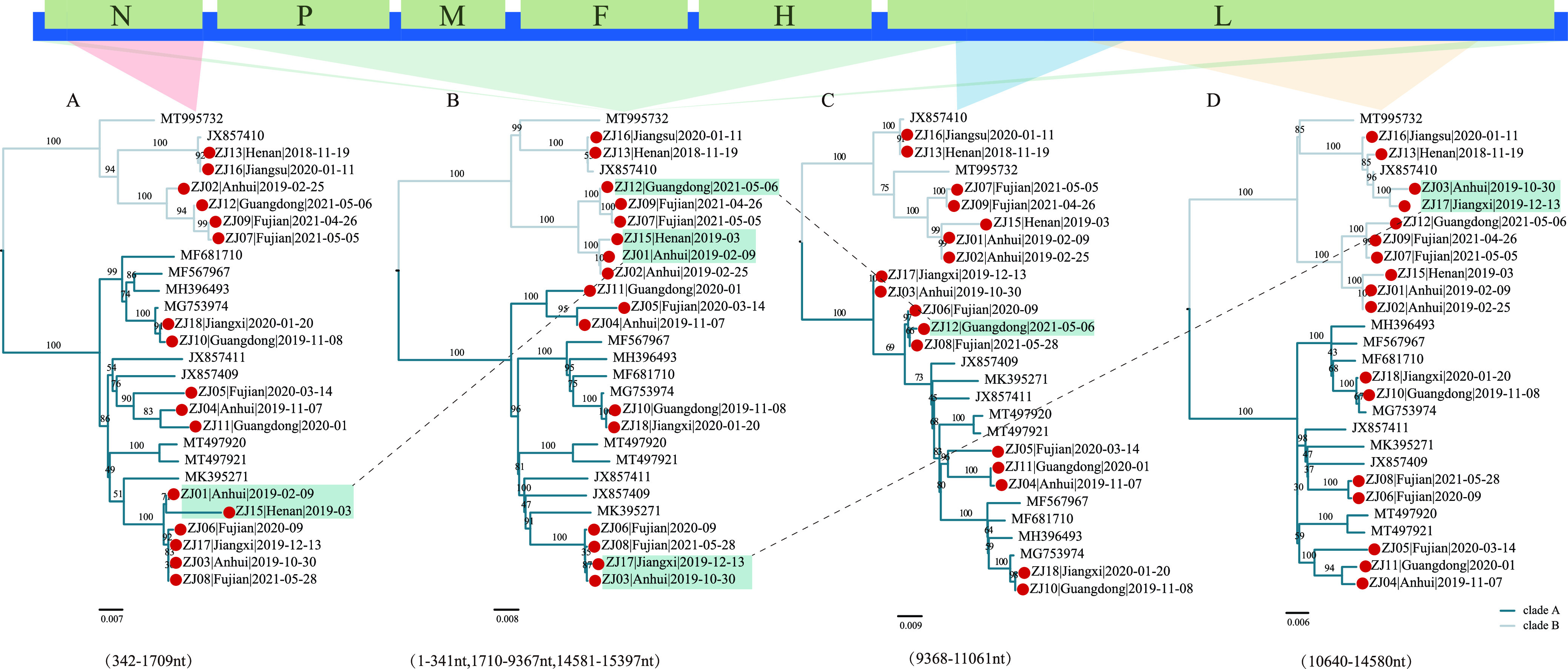
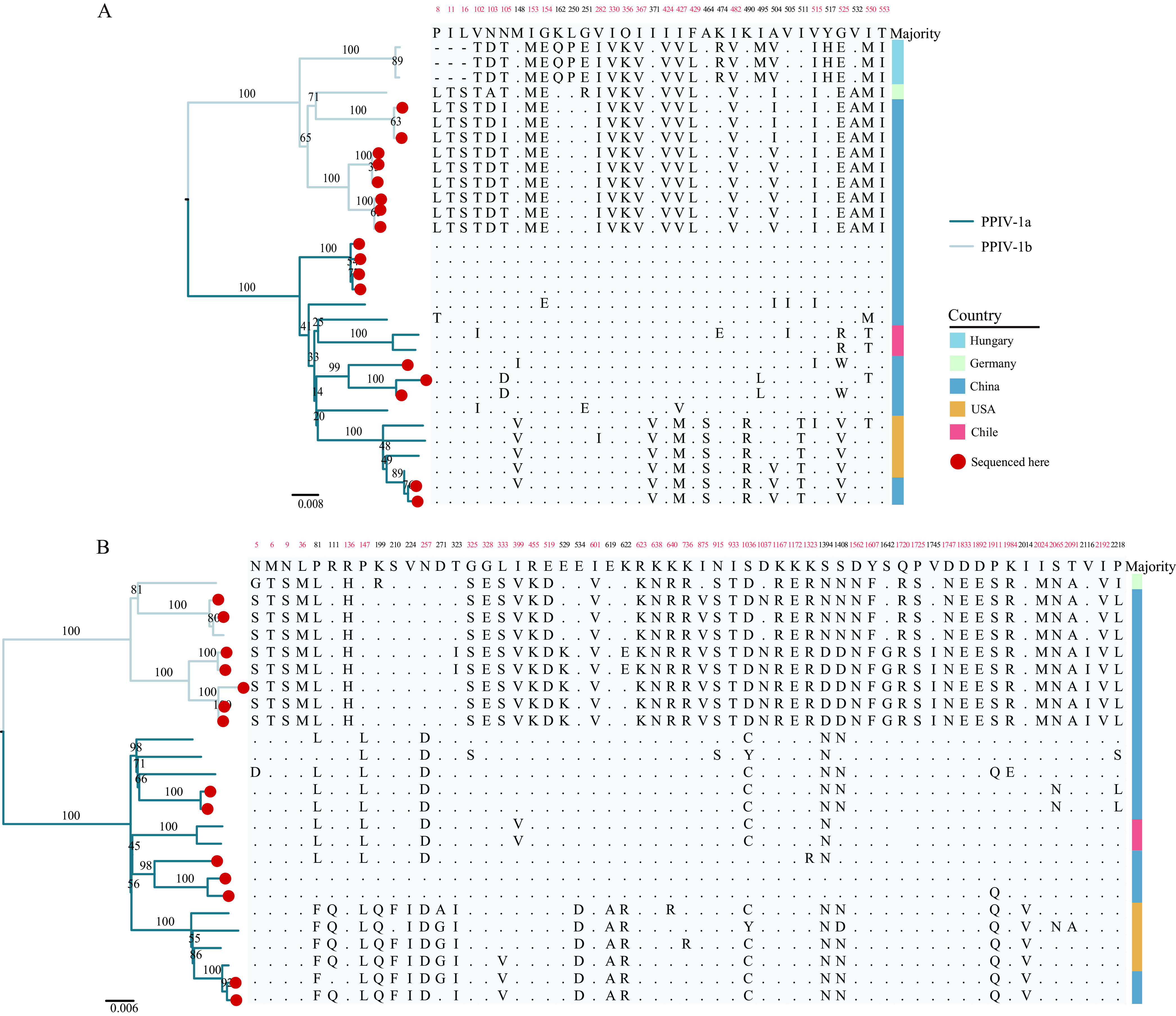
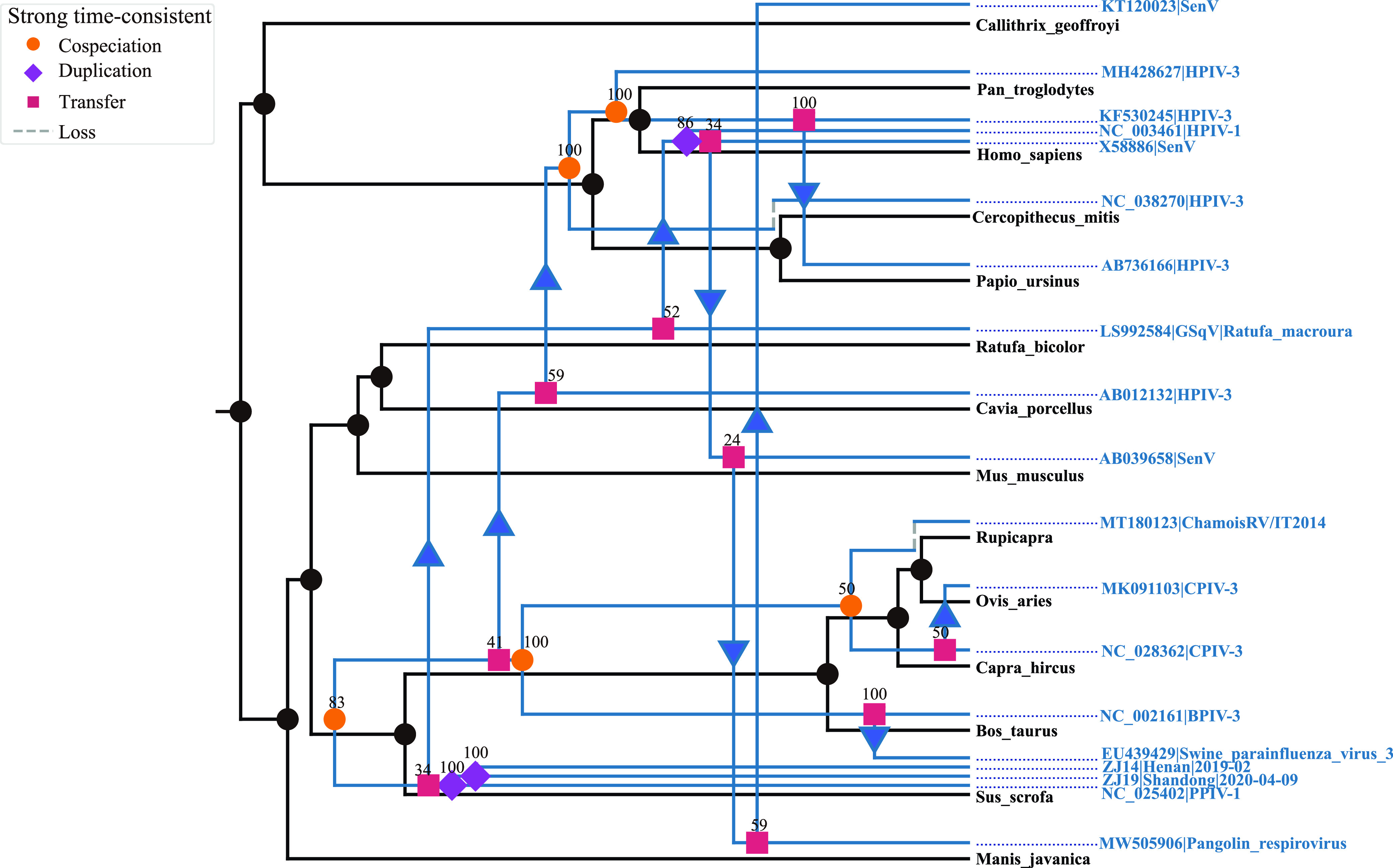
Paramyxoviridae is a rapidly growing family of viruses, whose potential for cross-species transmission makes it difficult to predict the harm of newly emerging viruses to humans and animals. To better understand their diversity, evolutionary history, and co-evolution with their hosts, we analyzed a collection of porcine parainfluenza virus (PPIV) genomes to reconstruct the species classification basis and evolutionary history of the Respirovirus genus. We sequenced 17 complete genomes of porcine respirovirus 1 (also known as porcine parainfluenza virus 1; PPIV-1), thereby nearly tripling the number of currently available PPIV-1 genomes. We found that PPIV-1 was widely prevalent in China with two divergent lineages, PPIV-1a and PPIV-1b. We further provided evidence that a new species, porcine parainfluenza virus 2 (PPIV-2), had recently emerged in China. Our results pointed to a need for revising the current species demarcation criteria of the Respirovirus genus. In addition, we used PPIV-1 as an example to explore recombination and diversity of the Respirovirus genus. Interestingly, we only detected heterosubtypic recombination events between PPIV-1a and PPIV-1b with no intrasubtypic recombination events. The recombination hotspots highlighted a diverse geography-dependent genome structure of paramyxovirus infecting swine in China. Furthermore, we found no evidence of co-evolution between respirovirus and its host, indicating frequent cross-species transmission. In summary, our analyses showed that swine can be infected with a broad range of respiroviruses and recombination may serve as an important evolutionary mechanism for the Respirovirus genus' greater diversity in genome structure than previously anticipated. IMPORTANCE Livestock have emerged as critically underrecognized sources of paramyxovirus diversity, including pigs serving as the source of Nipah virus (NiV) and swine parainfluenza virus type 3, and goats and bovines harboring highly divergent viral lineages. Here, we identified a new species of Respirovirus genus named PPIV-2 in swine and proposed to revise the species demarcation criteria of the Respirovirus genus. We found heterosubtypic recombination events and high genetic diversity in PPIV-1. Further, we showed that genetic recombination may have occurred in the Respirovirus genus which may be associated with host range expansion. The continued expansion of Respirovirus genus diversity in livestock with relatively high human contact rates requires enhanced surveillance and ongoing evaluation of emerging cross-species transmission threats.
Keywords: divergent viruses discovered; genetic diversity; porcine parainfluenza virus 2.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Identification and characterization of a novel paramyxovirus, porcine parainfluenza virus 1, from deceased pigs.J Gen Virol. 2013 Oct;94(Pt 10):2184-2190. doi: 10.1099/vir.0.052985-0. Epub 2013 Aug 5. J Gen Virol. 2013. PMID: 23918408
-
First report of porcine parainfluenza virus 1 (species Porcine respirovirus 1) in Europe.Transbound Emerg Dis. 2021 Jul;68(4):1731-1735. doi: 10.1111/tbed.13869. Epub 2020 Oct 17. Transbound Emerg Dis. 2021. PMID: 33006252
-
Detection of porcine parainfluenza virus type-1 antibody in swine serum using whole-virus ELISA, indirect fluorescence antibody and virus neutralizing assays.BMC Vet Res. 2022 Mar 21;18(1):110. doi: 10.1186/s12917-022-03196-6. BMC Vet Res. 2022. PMID: 35313864 Free PMC article.
-
Menangle and Nipah virus infections of pigs.Vet Clin North Am Food Anim Pract. 2002 Nov;18(3):557-71, ix. doi: 10.1016/s0749-0720(02)00038-5. Vet Clin North Am Food Anim Pract. 2002. PMID: 12442583 Review.
-
Exogenous porcine viruses.Curr Top Microbiol Immunol. 2003;278:125-83. doi: 10.1007/978-3-642-55541-1_6. Curr Top Microbiol Immunol. 2003. PMID: 12934944 Review.
Cited by
-
Zoonotic Paramyxoviruses: Evolution, Ecology, and Public Health Strategies in a Changing World.Viruses. 2024 Oct 29;16(11):1688. doi: 10.3390/v16111688. Viruses. 2024. Retraction in: Viruses. 2025 Jul 16;17(7):992. doi: 10.3390/v17070992. PMID: 39599803 Free PMC article. Retracted. Review.
-
Porcine Respirovirus 1 Suppresses Host Type I Interferon Production and the JAK-STAT Signaling Pathway.Viruses. 2023 May 16;15(5):1176. doi: 10.3390/v15051176. Viruses. 2023. PMID: 37243262 Free PMC article.
-
Genetic characterization of porcine parainfluenza virus 1 (PPIV-1) in pig farms: first report of PPIV-1 in Thailand and Myanmar.Front Vet Sci. 2025 Feb 19;12:1435920. doi: 10.3389/fvets.2025.1435920. eCollection 2025. Front Vet Sci. 2025. PMID: 40046421 Free PMC article.
-
Annual (2024) taxonomic update of RNA-directed RNA polymerase-encoding negative-sense RNA viruses (realm Riboviria: kingdom Orthornavirae: phylum Negarnaviricota).J Gen Virol. 2025 Jun;106(6):002077. doi: 10.1099/jgv.0.002077. J Gen Virol. 2025. PMID: 40512168 Free PMC article.
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
