

The longitudinal dynamics and natural history of clonal haematopoiesis
- PMID: 35650444
- PMCID: PMC9177423
- DOI: 10.1038/s41586-022-04785-z
The longitudinal dynamics and natural history of clonal haematopoiesis
Abstract
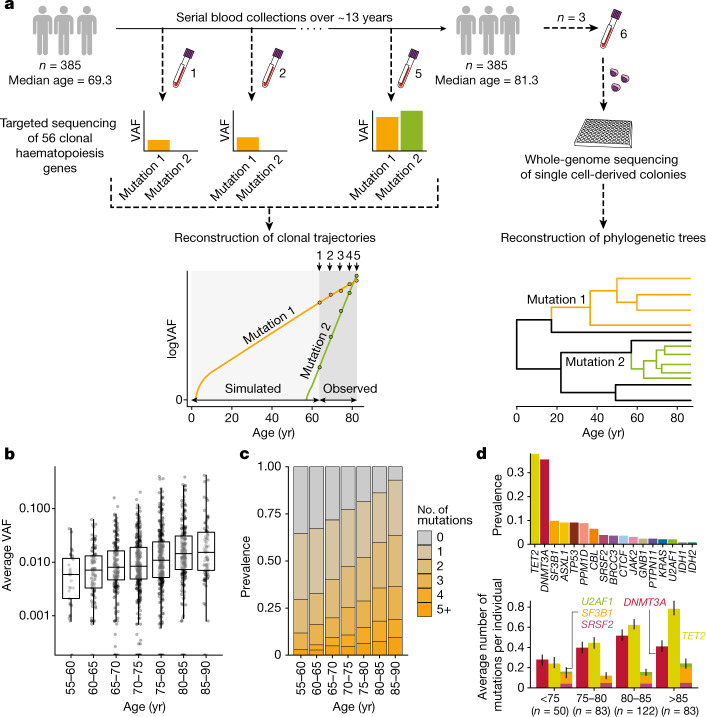
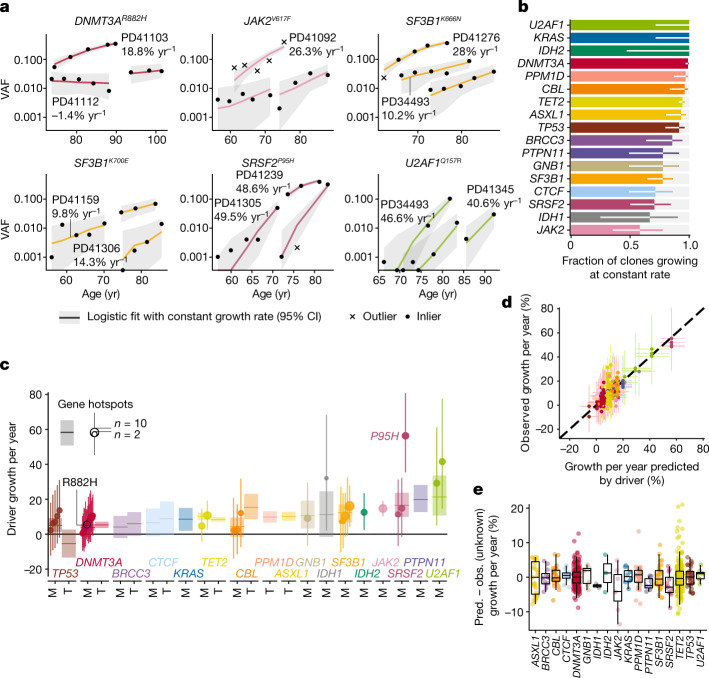
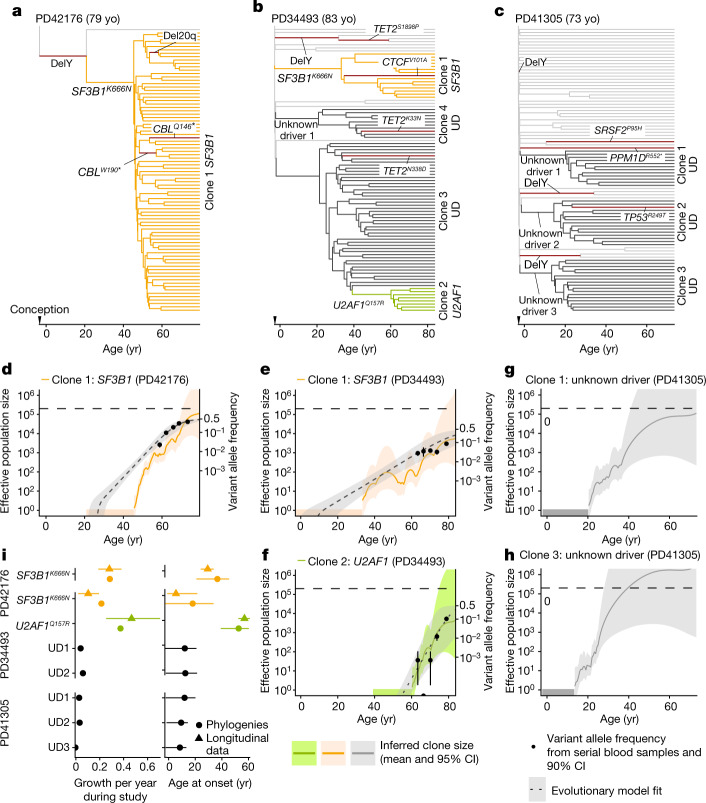
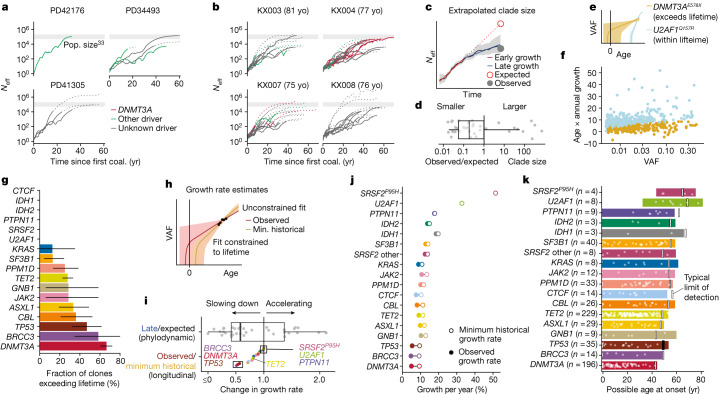
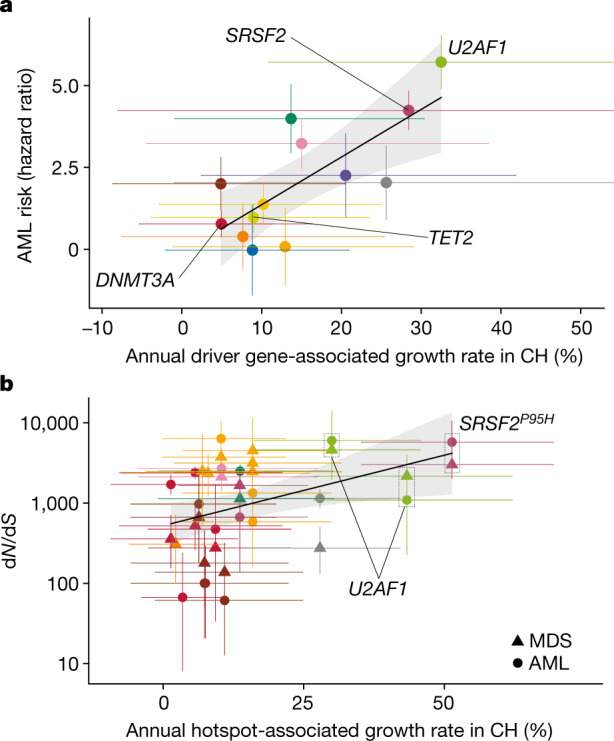
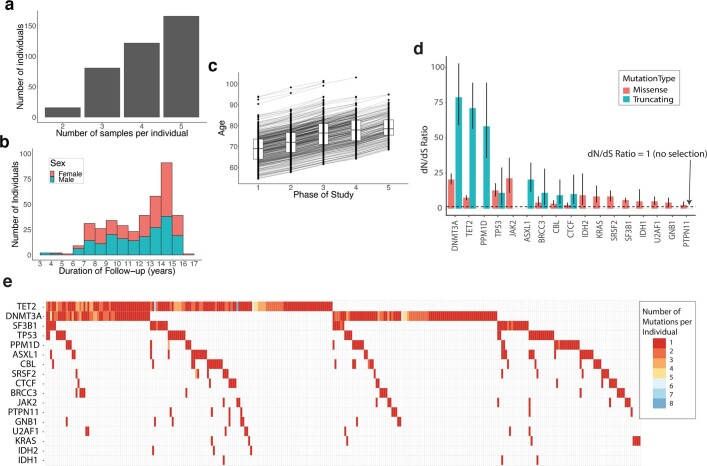
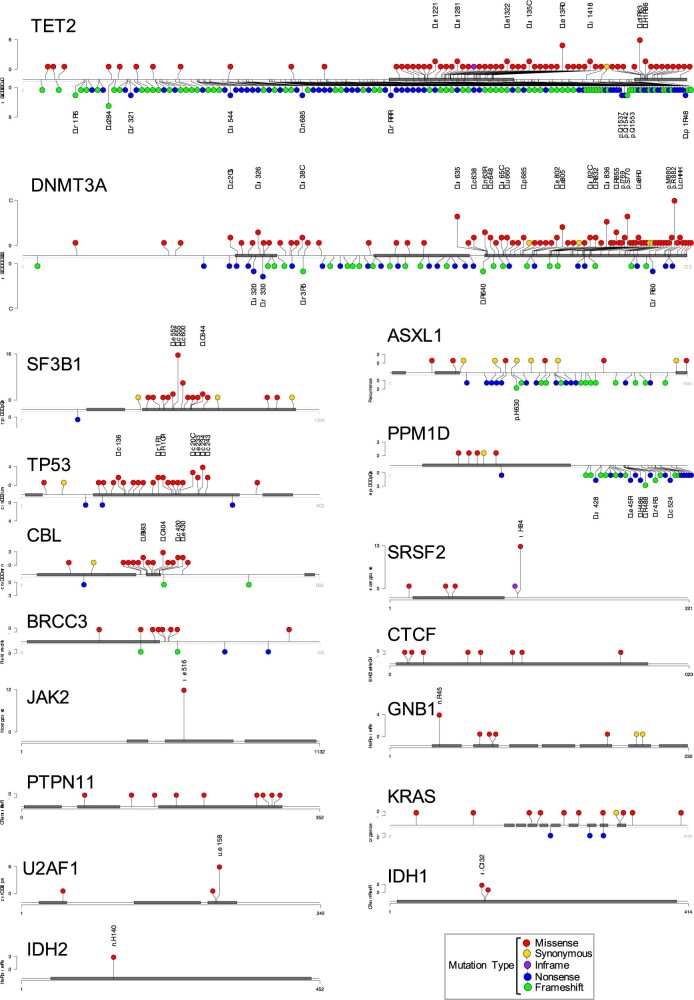
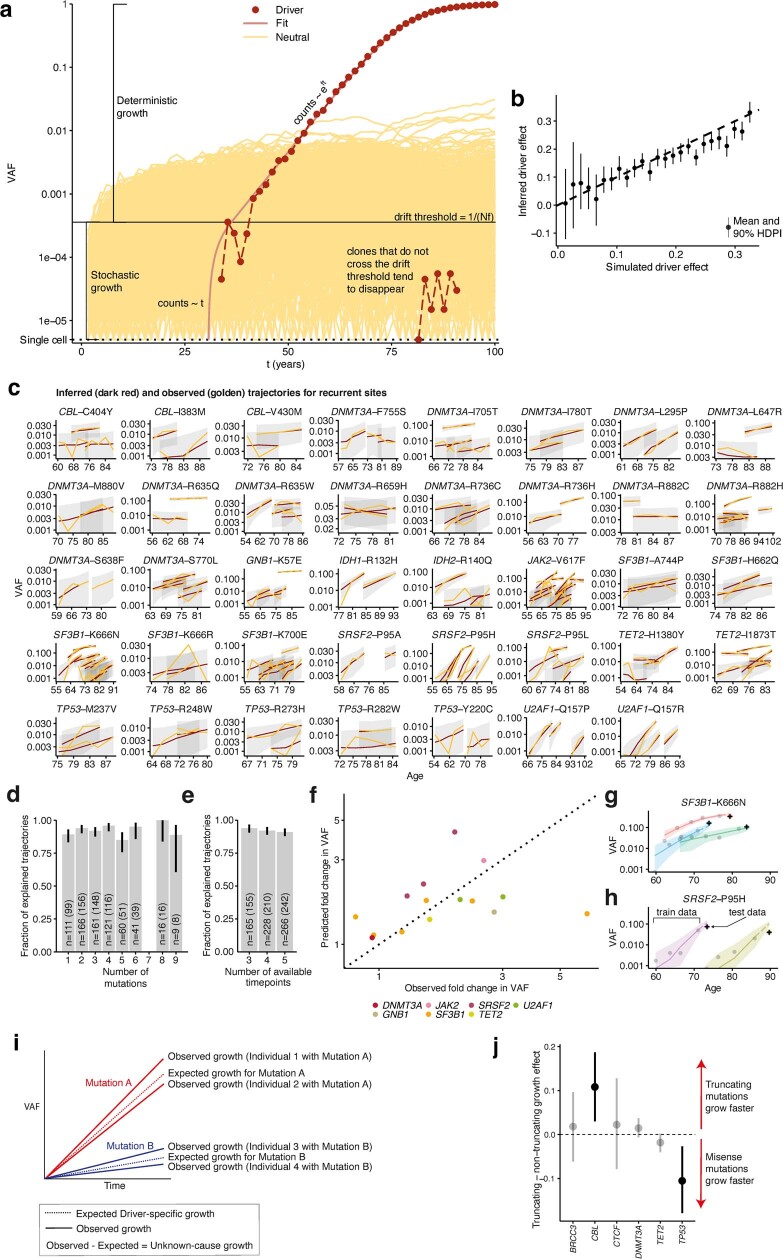
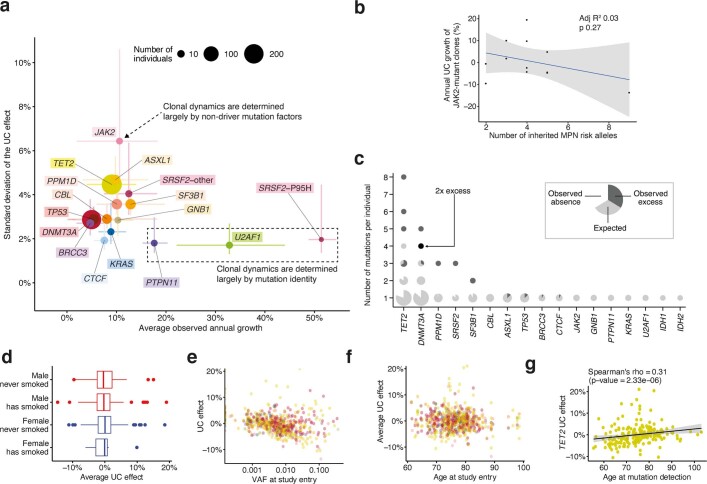
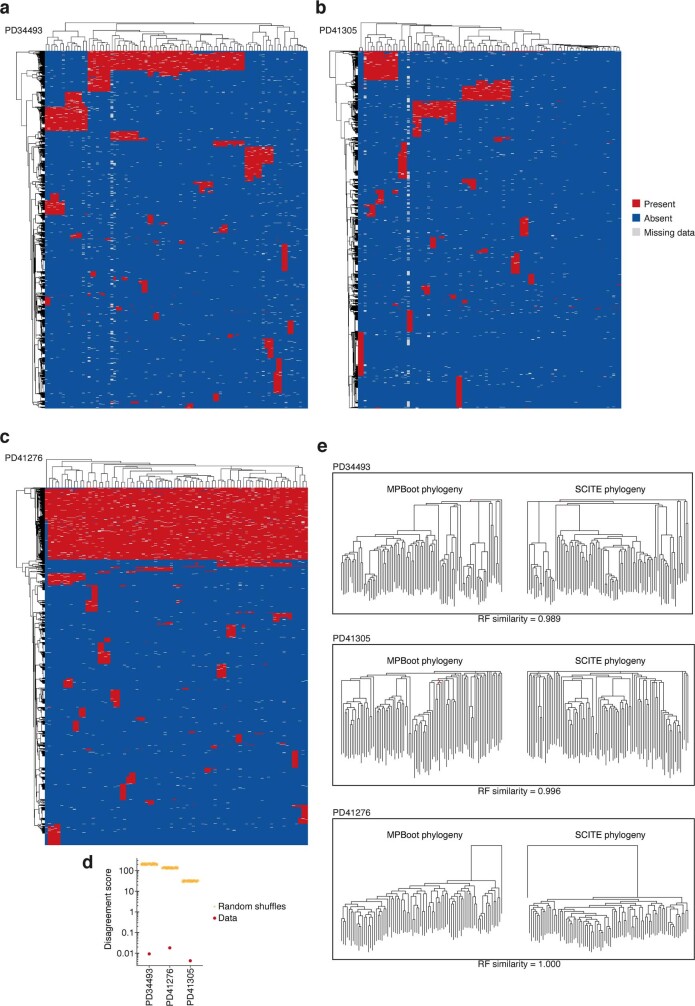
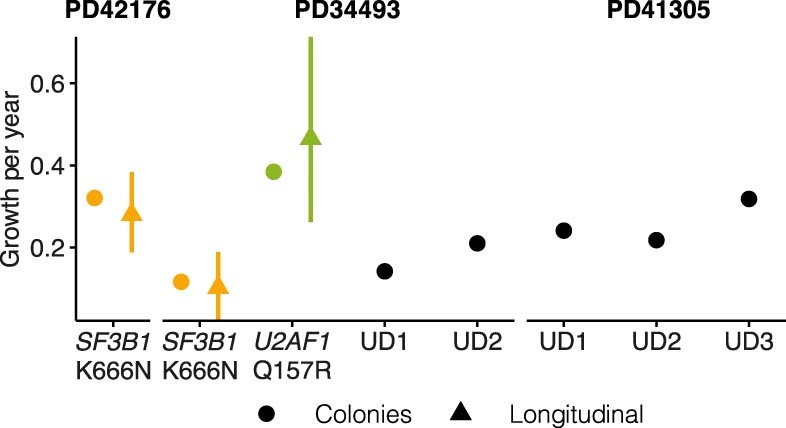
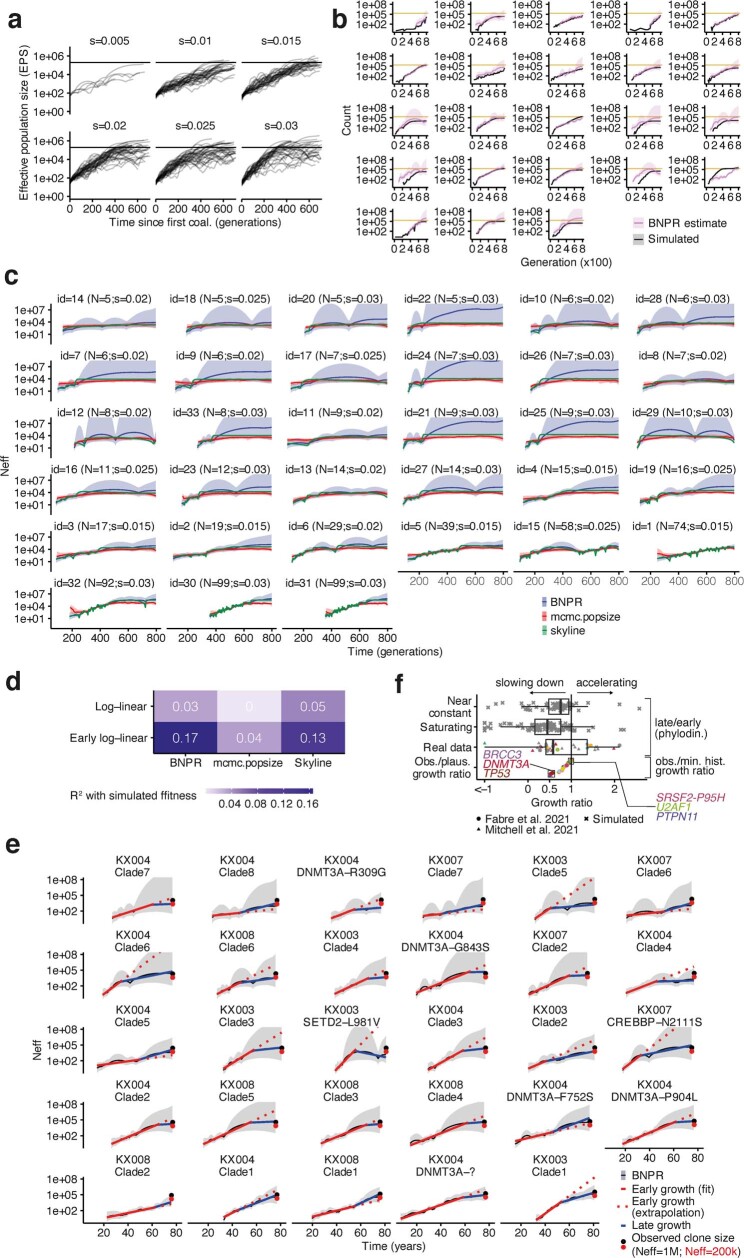
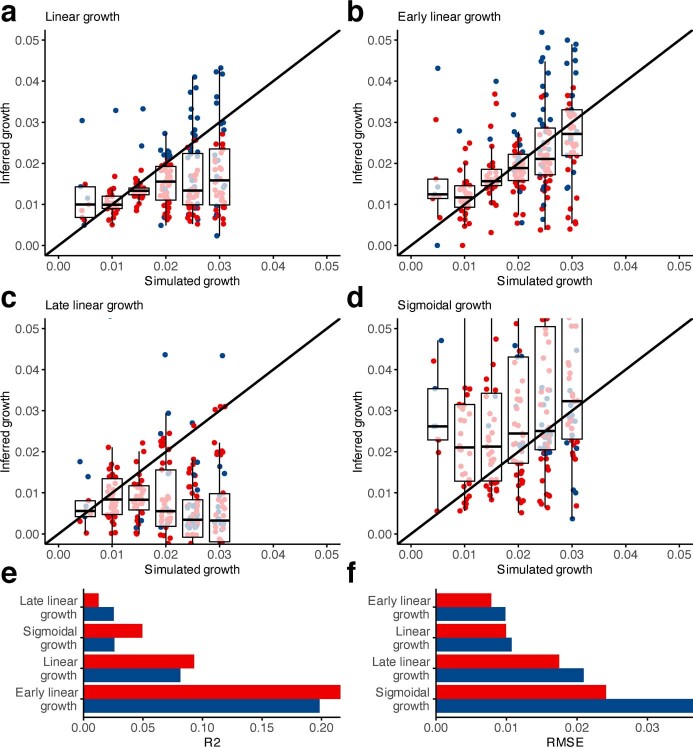
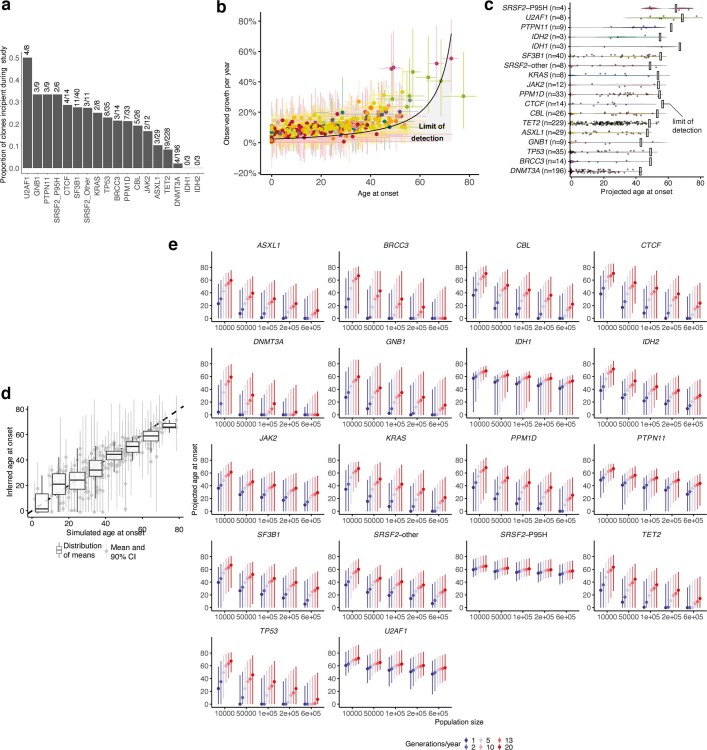
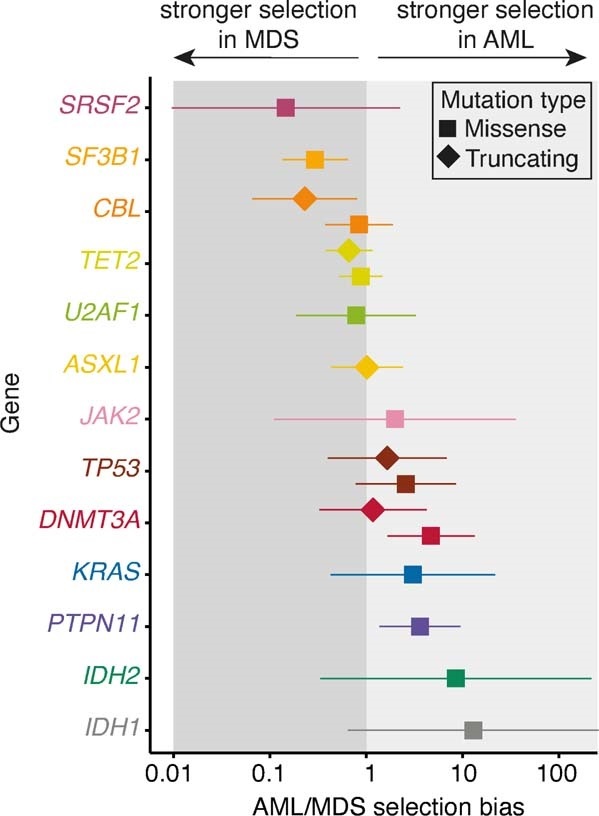
Clonal expansions driven by somatic mutations become pervasive across human tissues with age, including in the haematopoietic system, where the phenomenon is termed clonal haematopoiesis1-4. The understanding of how and when clonal haematopoiesis develops, the factors that govern its behaviour, how it interacts with ageing and how these variables relate to malignant progression remains limited5,6. Here we track 697 clonal haematopoiesis clones from 385 individuals 55 years of age or older over a median of 13 years. We find that 92.4% of clones expanded at a stable exponential rate over the study period, with different mutations driving substantially different growth rates, ranging from 5% (DNMT3A and TP53) to more than 50% per year (SRSF2P95H). Growth rates of clones with the same mutation differed by approximately ±5% per year, proportionately affecting slow drivers more substantially. By combining our time-series data with phylogenetic analysis of 1,731 whole-genome sequences of haematopoietic colonies from 7 individuals from an older age group, we reveal distinct patterns of lifelong clonal behaviour. DNMT3A-mutant clones preferentially expanded early in life and displayed slower growth in old age, in the context of an increasingly competitive oligoclonal landscape. By contrast, splicing gene mutations drove expansion only later in life, whereas TET2-mutant clones emerged across all ages. Finally, we show that mutations driving faster clonal growth carry a higher risk of malignant progression. Our findings characterize the lifelong natural history of clonal haematopoiesis and give fundamental insights into the interactions between somatic mutation, ageing and clonal selection.
© 2022. The Author(s).
Conflict of interest statement
G.S.V. is a consultant for STRM.BIO and receives a research grant from Astrazeneca. The other authors declare no competing interests.
Figures
Comment in
-
Blood's life history traced through genomic scars.Nature. 2022 Jun;606(7913):255-256. doi: 10.1038/d41586-022-01304-y. Nature. 2022. PMID: 35650394 No abstract available.
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
