

Fabry Disease: Current and Novel Therapeutic Strategies. A Narrative Review
- PMID: 35652398
- PMCID: PMC10207921
- DOI: 10.2174/1570159X20666220601124117
Fabry Disease: Current and Novel Therapeutic Strategies. A Narrative Review
Abstract
Background: Fabry disease (FD) is an inherited lysosomal storage disorder, leading to multisystemic manifestations and causing significant morbidity and mortality.
Objective: The aim of this narrative review is to present the current and novel therapeutic strategies in FD, including symptomatic and specific treatment options.
Methods: A systematic literature search was conducted to identify relevant studies, including completed and ongoing randomized-controlled clinical trials (RCTs), prospective or retrospective cohort studies, case series and case reports that provided clinical data regarding FD treatment.
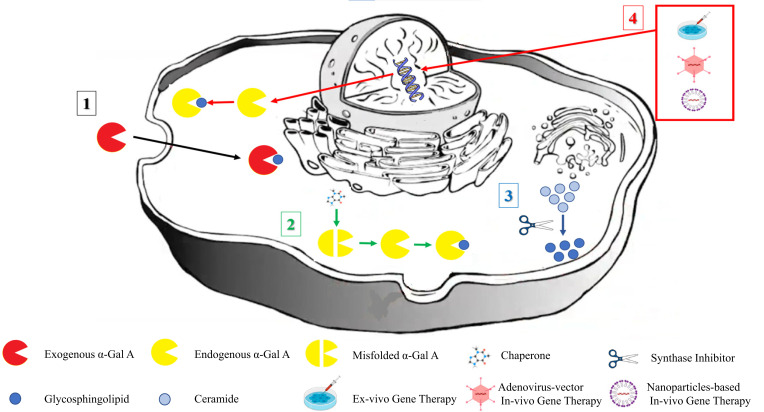
Results: A multidisciplinary symptomatic treatment is recommended for FD patients, personalized according to disease manifestations and their severity. During the last two decades, FD-specific treatments, including two enzyme-replacement-therapies (agalsidase alfa and agalsidase beta) and chaperone treatment with migalastat have been approved for use and allowed for symptoms' stabilization or even disease burden reduction. More therapeutic agents are currently under investigation. Substrate reduction therapies, including lucerastat and venglustat, have shown promising results in RCTs and may be used either as monotherapy or as complementary therapy to established enzymereplacement- therapies. More stable enzyme-replacement-therapy molecules that are associated with less adverse events and lower likelihood of neutralizing antibodies formation have also been developed. Ex-vivo and in-vivo gene therapy is being tested in animal models and pilot human clinical trials, with preliminary results showing a favorable safety and efficacy profile.
Conclusion: The therapeutic landscape in FD appears to be actively expanding with more treatment options expected to become available in the near future, allowing for a more personalized approach in FD patients.
Keywords: Fabry disease; chaperone; enzyme replacement therapy; gene therapy; mutation; rare neurological diseases.
Copyright© Bentham Science Publishers; For any queries, please email at epub@benthamscience.net.
Conflict of interest statement
The authors declare no conflict of interest, financial or otherwise.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
