

A mitochondrial unfolded protein response inhibitor suppresses prostate cancer growth in mice via HSP60
- PMID: 35653190
- PMCID: PMC9246382
- DOI: 10.1172/JCI149906
A mitochondrial unfolded protein response inhibitor suppresses prostate cancer growth in mice via HSP60
Abstract
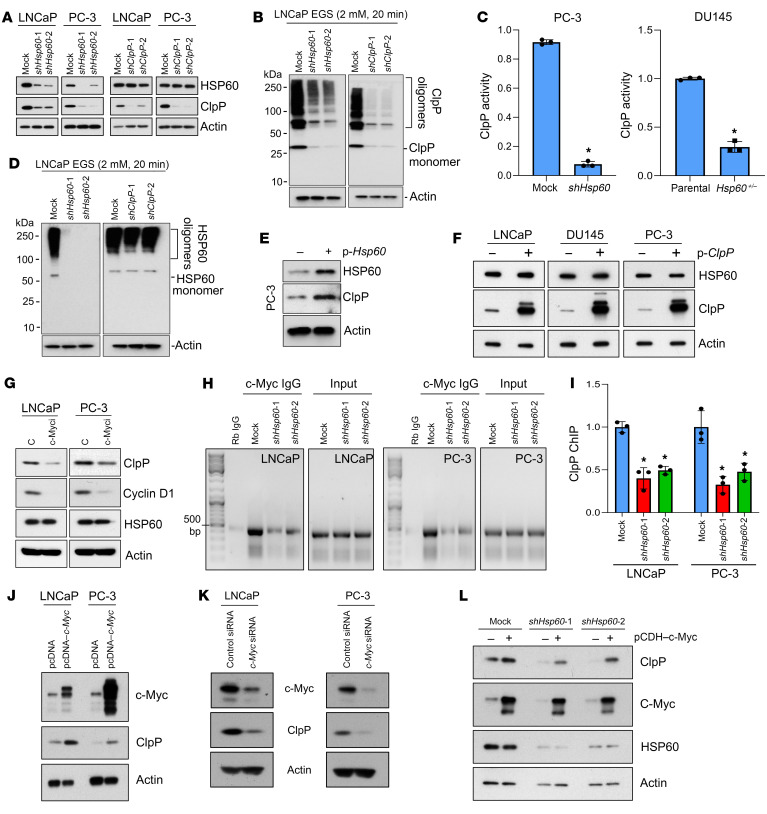
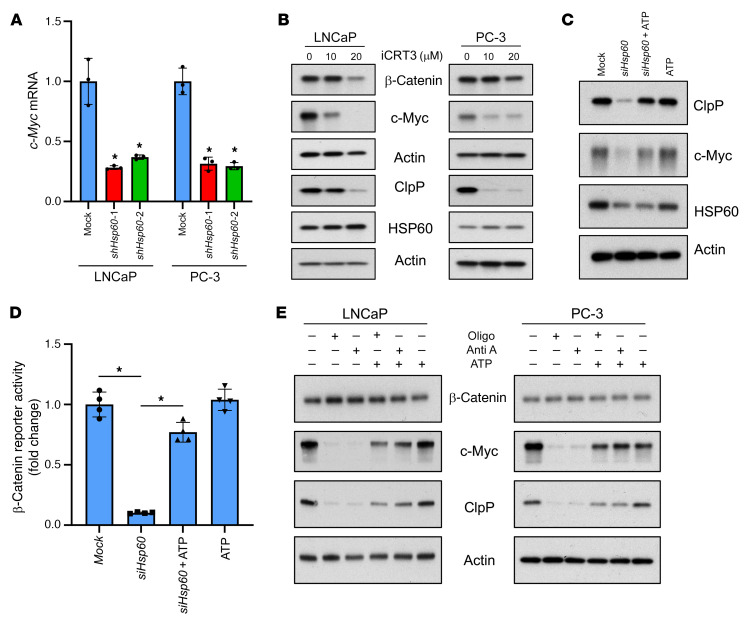
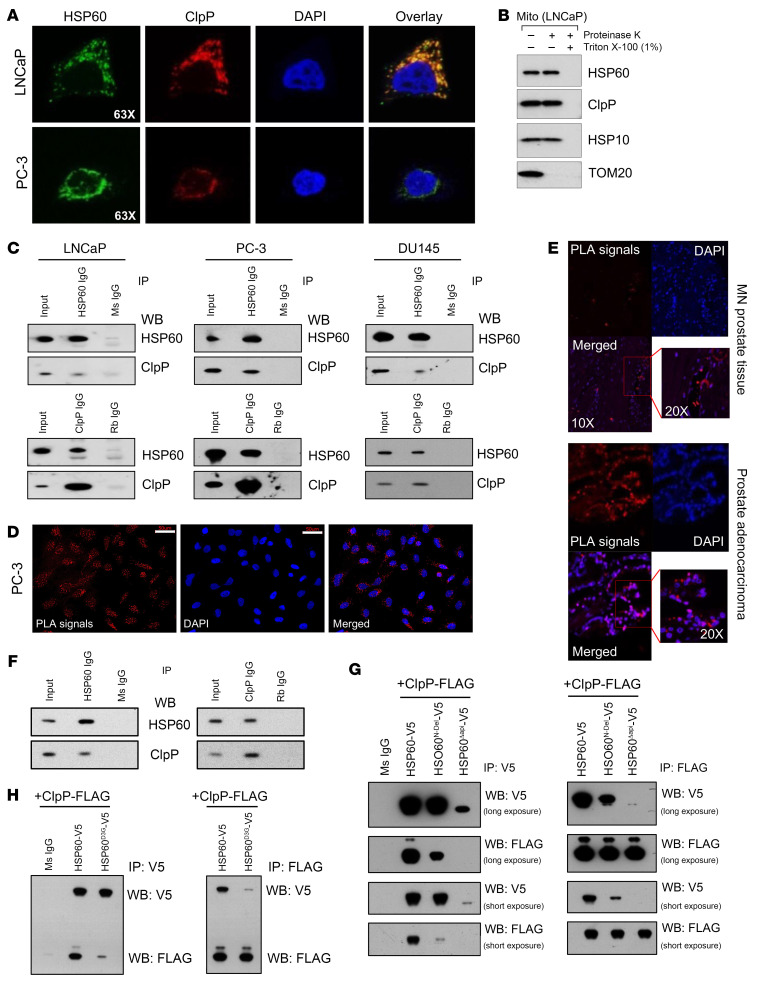
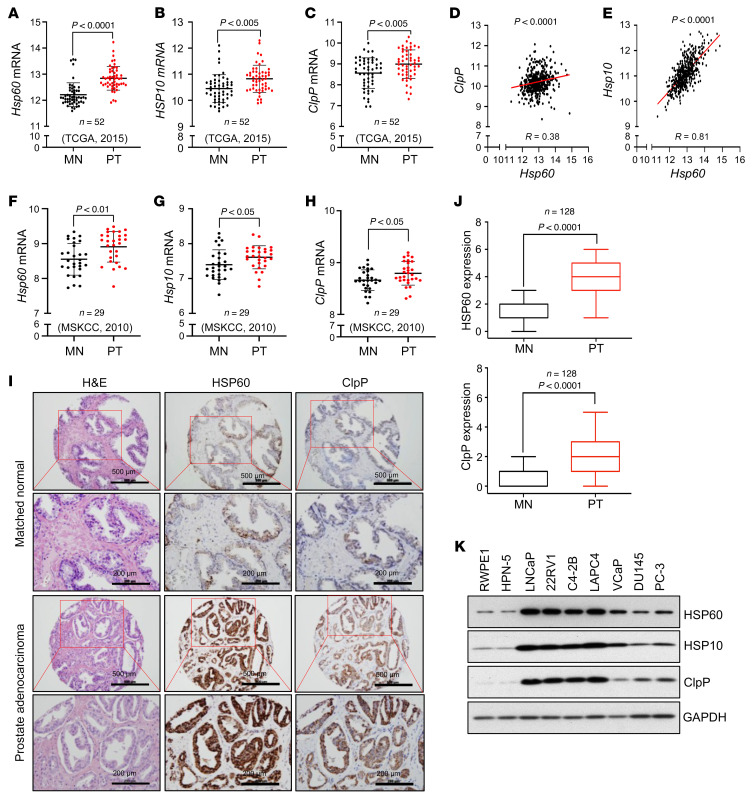
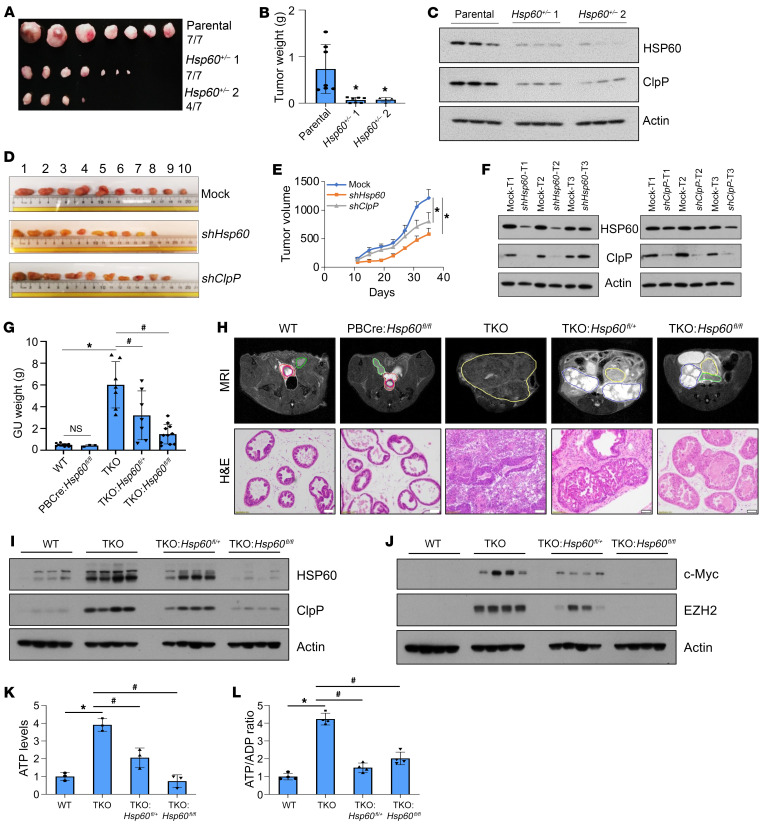
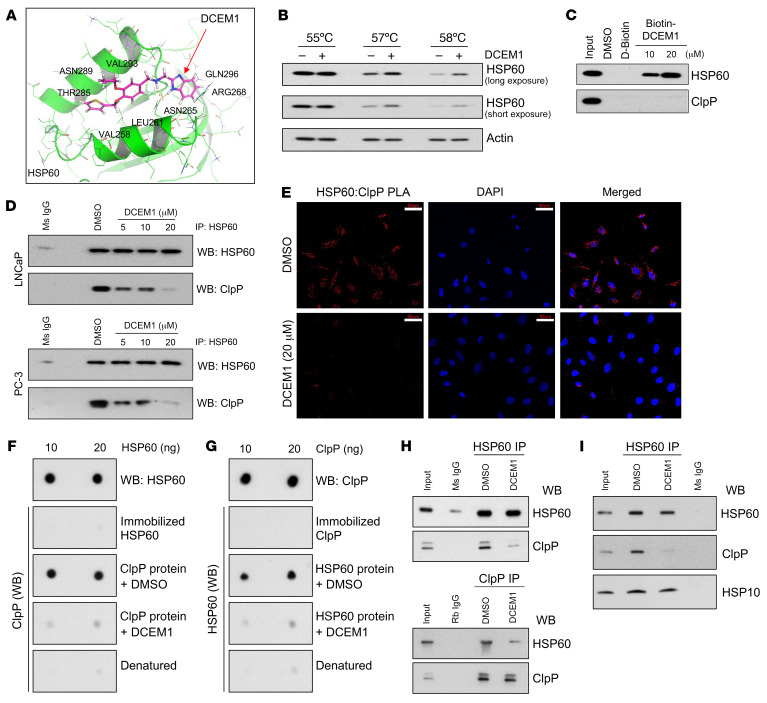
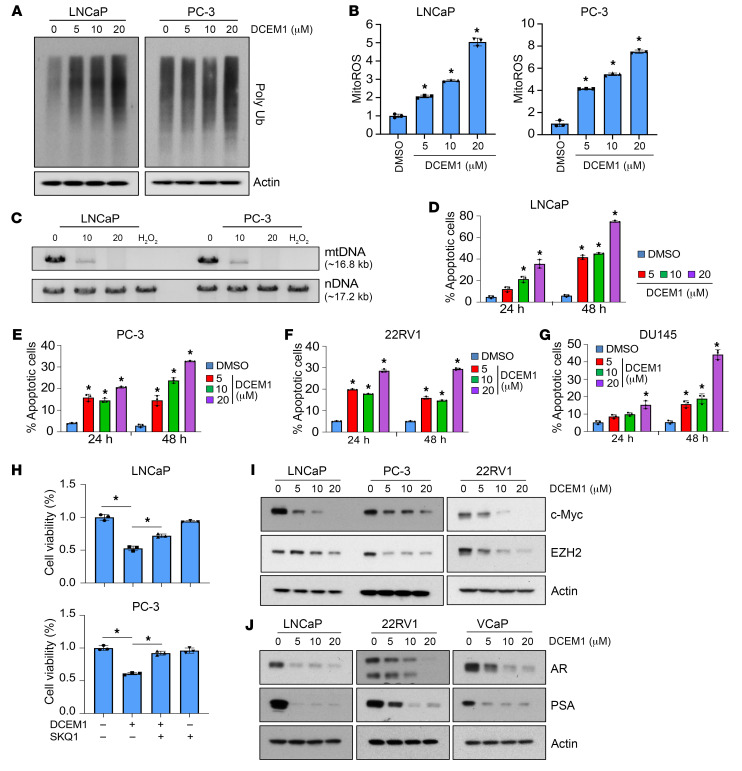
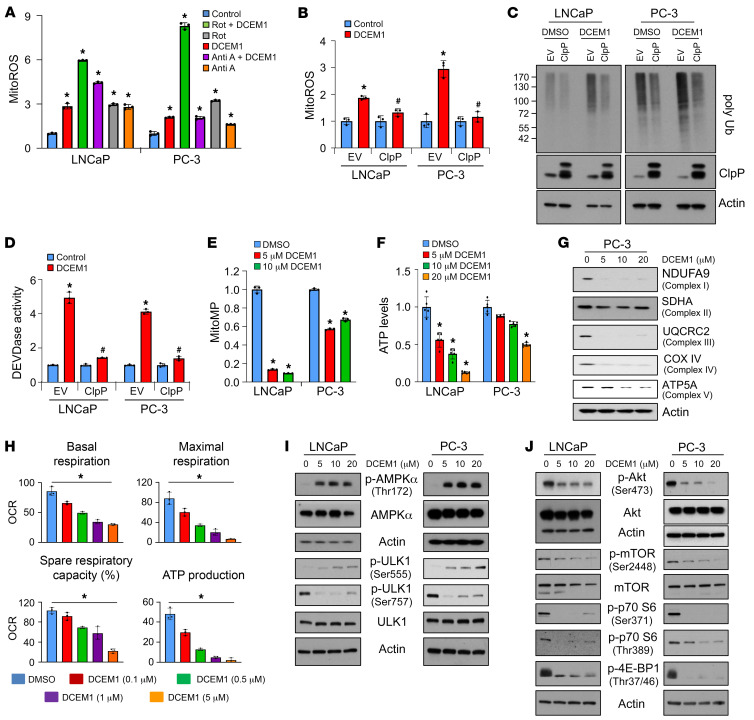
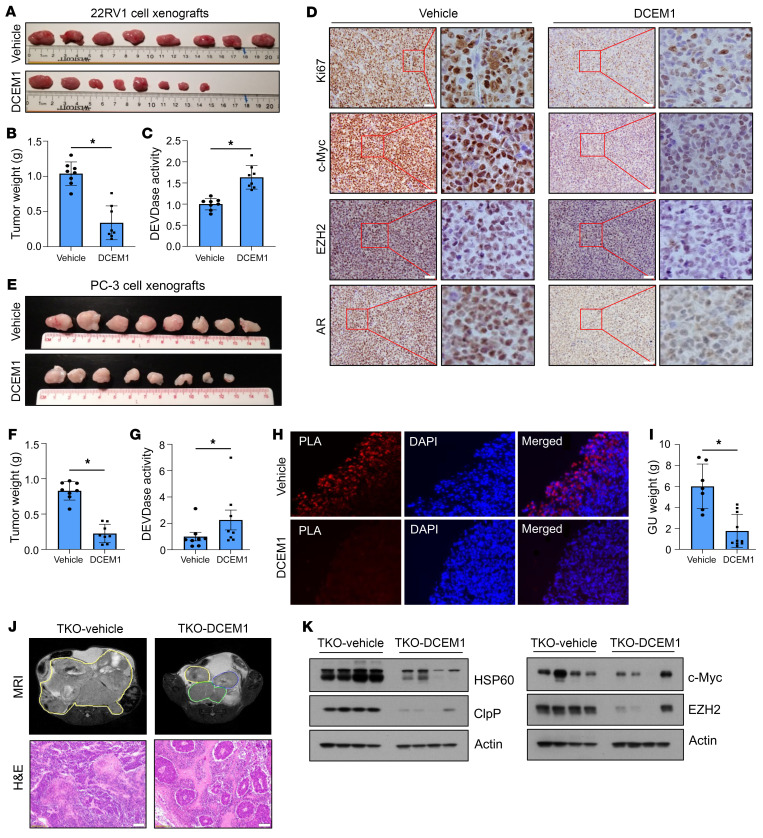
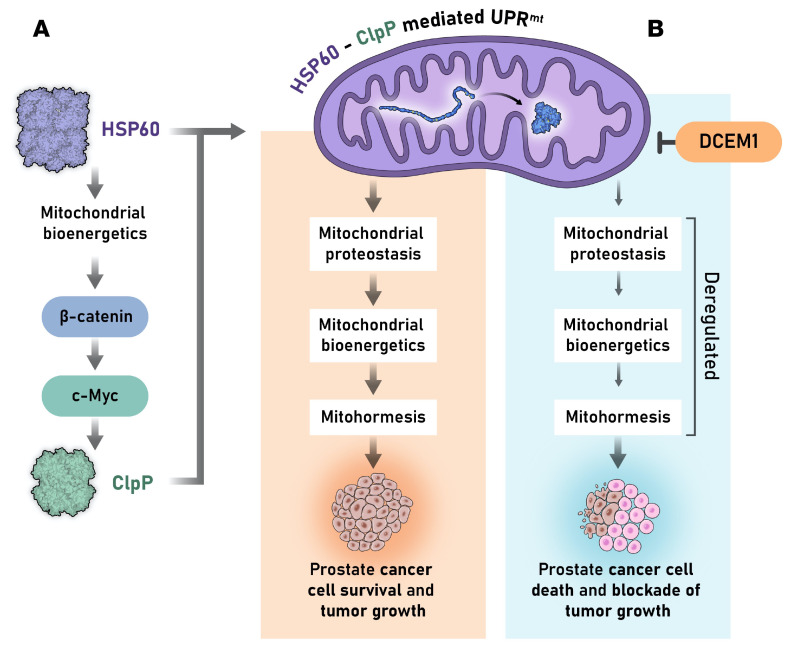
Mitochondrial proteostasis, regulated by the mitochondrial unfolded protein response (UPRmt), is crucial for maintenance of cellular functions and survival. Elevated oxidative and proteotoxic stress in mitochondria must be attenuated by the activation of a ubiquitous UPRmt to promote prostate cancer (PCa) growth. Here we show that the 2 key components of the UPRmt, heat shock protein 60 (HSP60, a mitochondrial chaperonin) and caseinolytic protease P (ClpP, a mitochondrial protease), were required for the development of advanced PCa. HSP60 regulated ClpP expression via c-Myc and physically interacted with ClpP to restore mitochondrial functions that promote cancer cell survival. HSP60 maintained the ATP-producing functions of mitochondria, which activated the β-catenin pathway and led to the upregulation of c-Myc. We identified a UPRmt inhibitor that blocked HSP60's interaction with ClpP and abrogated survival signaling without altering HSP60's chaperonin function. Disruption of HSP60-ClpP interaction with the UPRmt inhibitor triggered metabolic stress and impeded PCa-promoting signaling. Treatment with the UPRmt inhibitor or genetic ablation of Hsp60 inhibited PCa growth and progression. Together, our findings demonstrate that the HSP60-ClpP-mediated UPRmt is essential for prostate tumorigenesis and the HSP60-ClpP interaction represents a therapeutic vulnerability in PCa.
Keywords: Cell Biology; Cell stress; Mitochondria; Oncology; Prostate cancer.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
