

Recurrent noncoding somatic and germline WT1 variants converge to disrupt MYB binding in acute promyelocytic leukemia
- PMID: 35653587
- PMCID: PMC9461475
- DOI: 10.1182/blood.2021014945
Recurrent noncoding somatic and germline WT1 variants converge to disrupt MYB binding in acute promyelocytic leukemia
Abstract
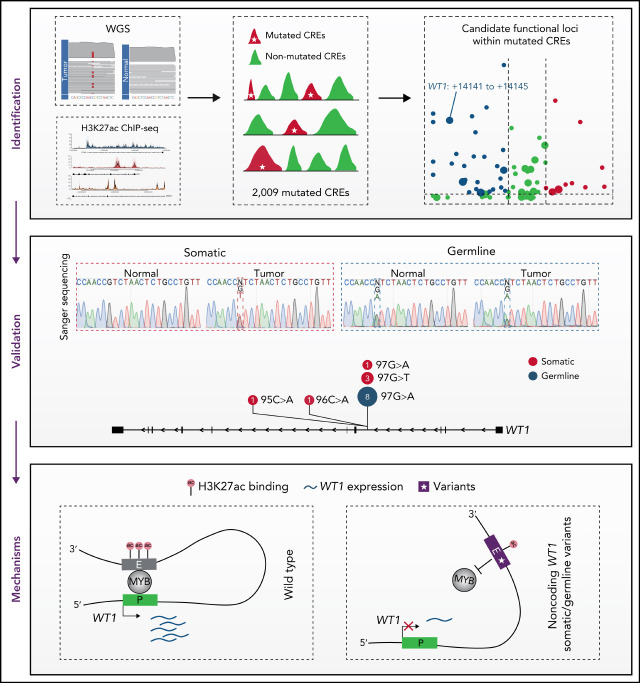
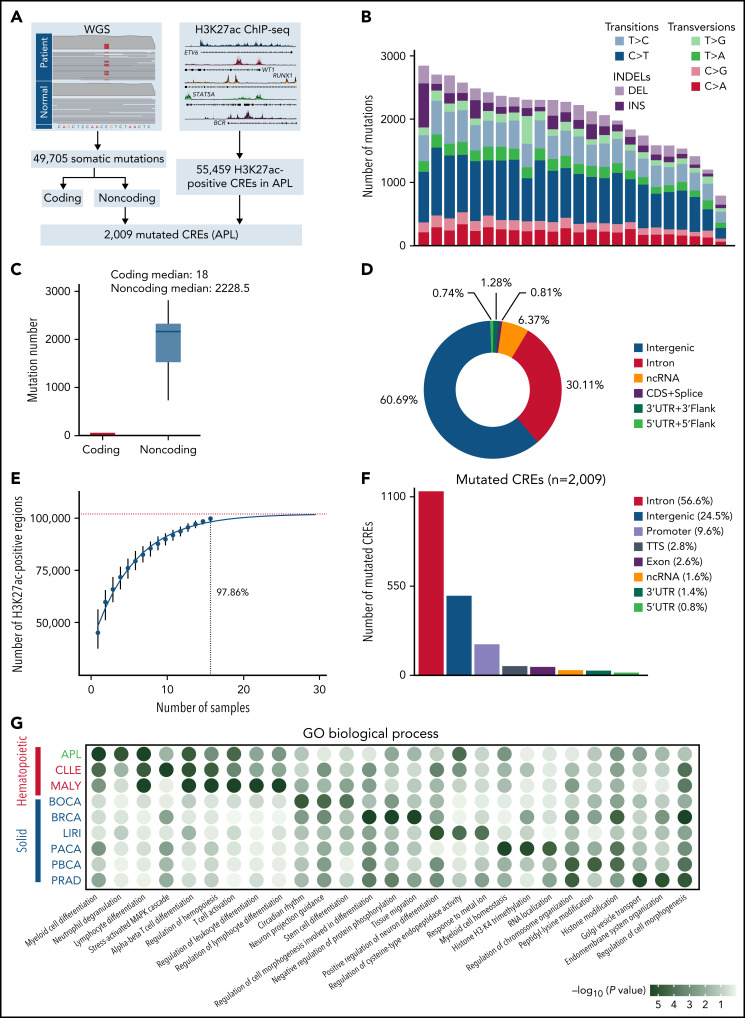
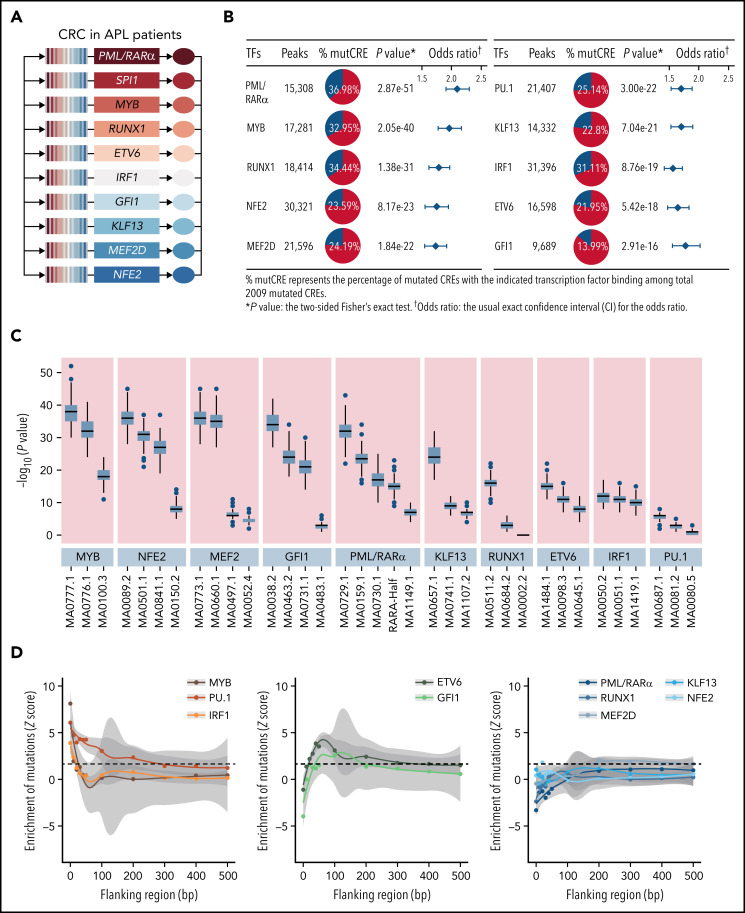
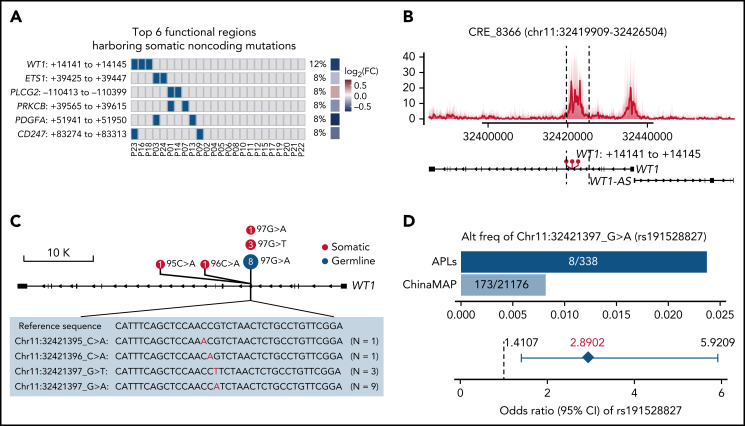
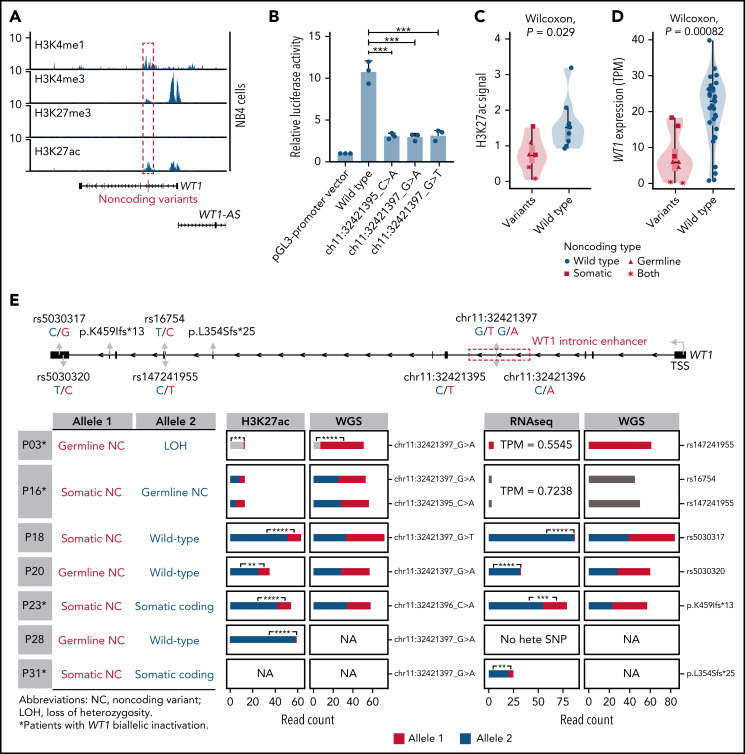
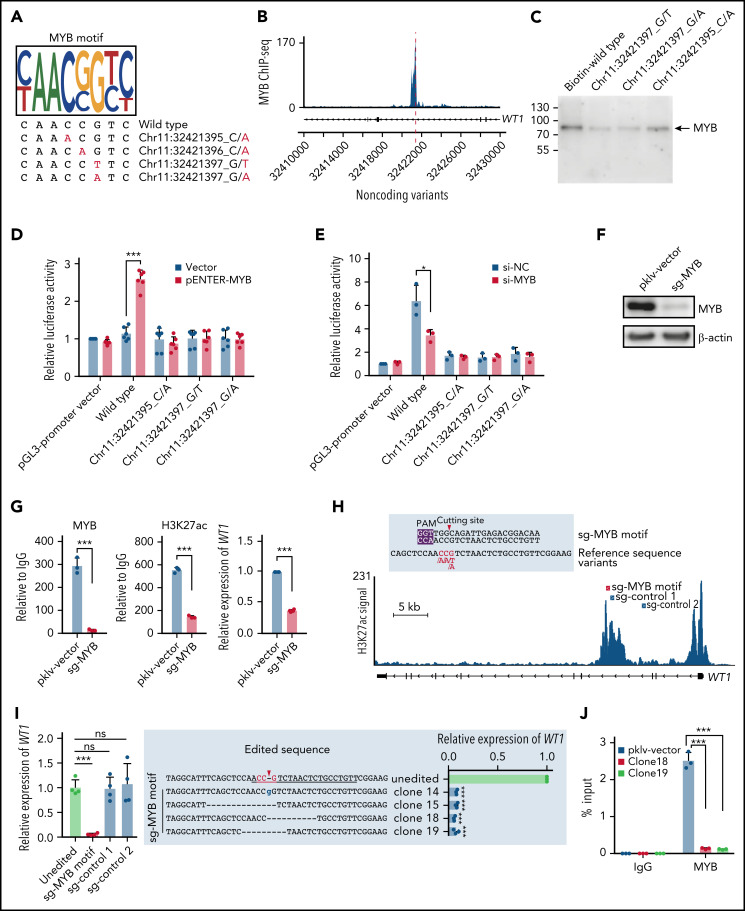
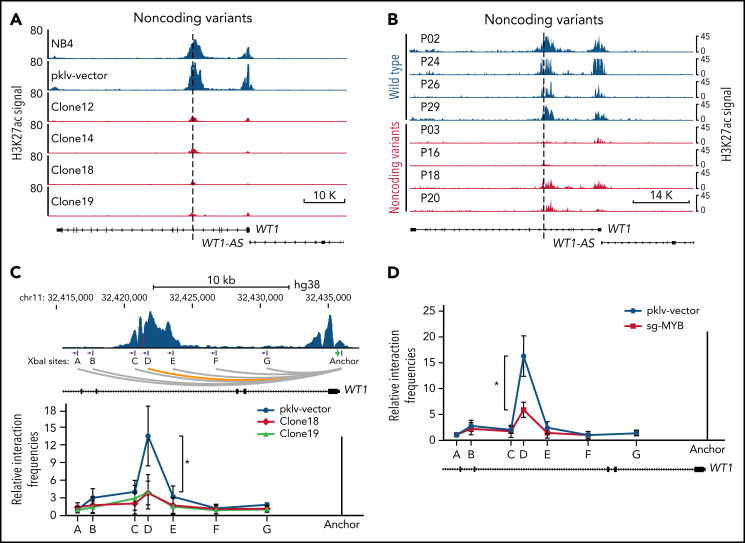
Genetic alternations can occur at noncoding regions, but how they contribute to cancer pathogenesis is poorly understood. Here, we established a mutational landscape of cis-regulatory regions (CREs) in acute promyelocytic leukemia (APL) based on whole-genome sequencing analysis of paired tumor and germline samples from 24 patients and epigenetic profiling of 16 patients. Mutations occurring in CREs occur preferentially in active enhancers bound by the complex of master transcription factors in APL. Among significantly enriched mutated CREs, we found a recurrently mutated region located within the third intron of WT1, an essential regulator of normal and malignant hematopoiesis. Focusing on noncoding mutations within this WT1 intron, an analysis on 169 APL patients revealed that somatic mutations were clustered into a focal hotspot region, including one site identified as a germline polymorphism contributing to APL risk. Significantly decreased WT1 expression was observed in APL patients bearing somatic and/or germline noncoding WT1 variants. Furthermore, biallelic WT1 inactivation was recurrently found in APL patients with noncoding WT1 variants, which resulted in the complete loss of WT1. The high incidence of biallelic inactivation suggested the tumor suppressor activity of WT1 in APL. Mechanistically, noncoding WT1 variants disrupted MYB binding on chromatin and suppressed the enhancer activity and WT1 expression through destroying the chromatin looping formation. Our study highlights the important role of noncoding variants in the leukemogenesis of APL.
© 2022 by The American Society of Hematology.
Figures
Comment in
-
In APL, noncoding mutations and SNP converge on WT1.Blood. 2022 Sep 8;140(10):1060-1061. doi: 10.1182/blood.2022017214. Blood. 2022. PMID: 36074531 No abstract available.
References
-
- Mazrooei P, Kron KJ, Zhu Y, et al. . Cistrome partitioning reveals convergence of somatic mutations and risk variants on master transcription regulators in primary prostate tumors. Cancer Cell. 2019;36(6):674-689.e6. - PubMed
-
- Li J, Zhang T, Ramakrishnan A, et al. . Dynamic changes in cis-regulatory occupancy by Six1 and its cooperative interactions with distinct cofactors drive lineage-specific gene expression programs during progressive differentiation of the auditory sensory epithelium. Nucleic Acids Res. 2020;48(6):2880-2896. - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
