

Identification of cell types in multiplexed in situ images by combining protein expression and spatial information using CELESTA
- PMID: 35654951
- PMCID: PMC9728133
- DOI: 10.1038/s41592-022-01498-z
Identification of cell types in multiplexed in situ images by combining protein expression and spatial information using CELESTA
Abstract
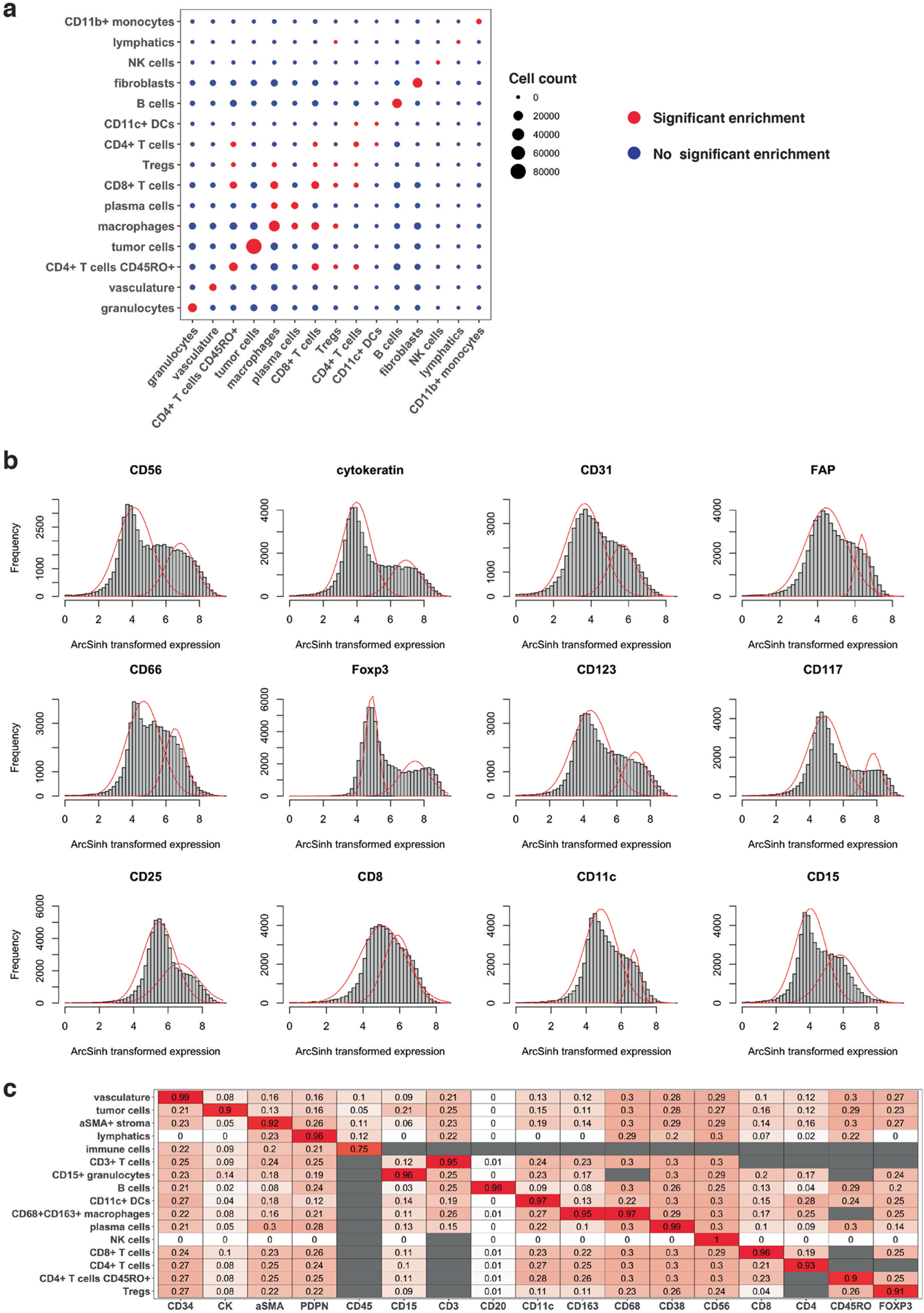
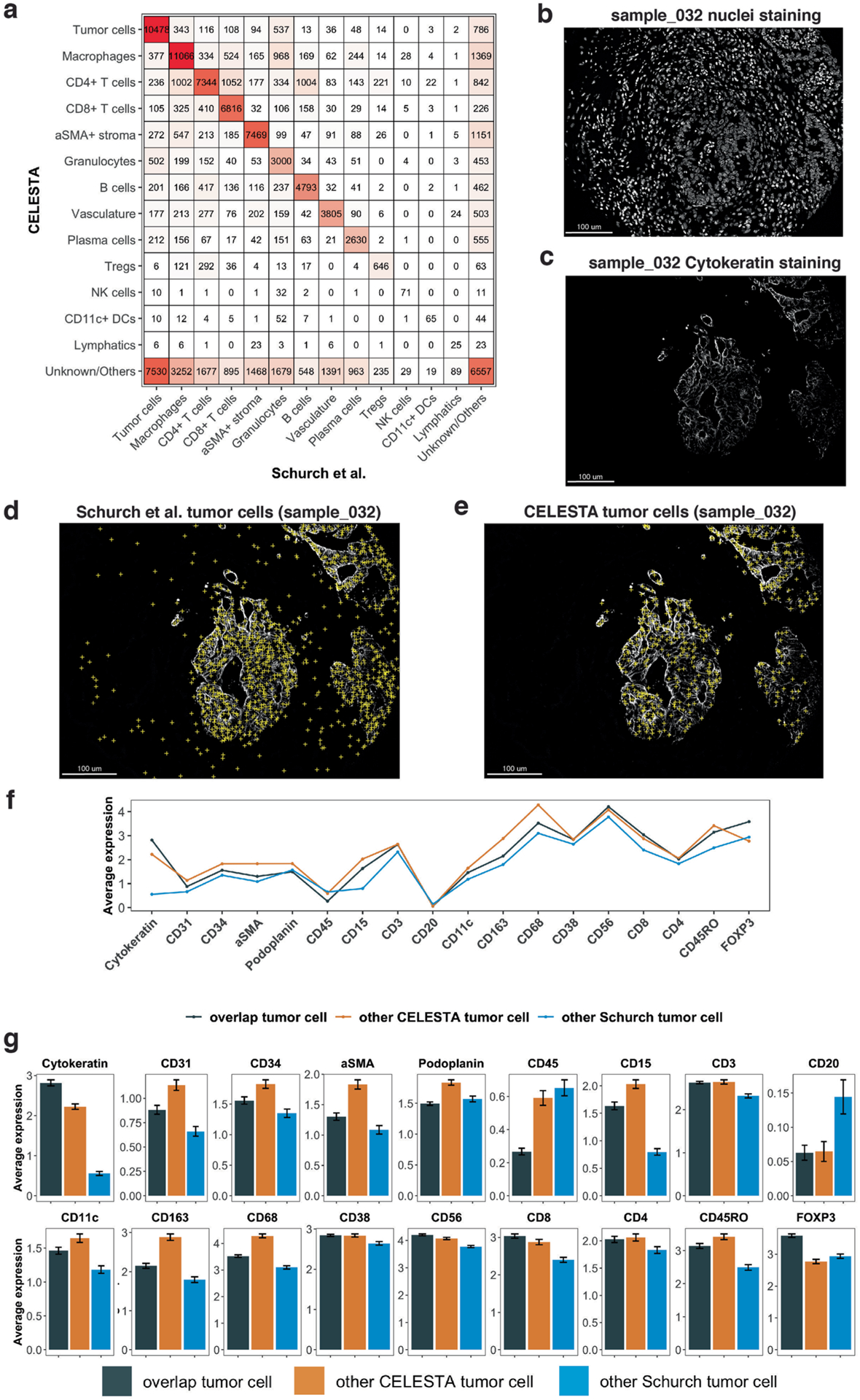
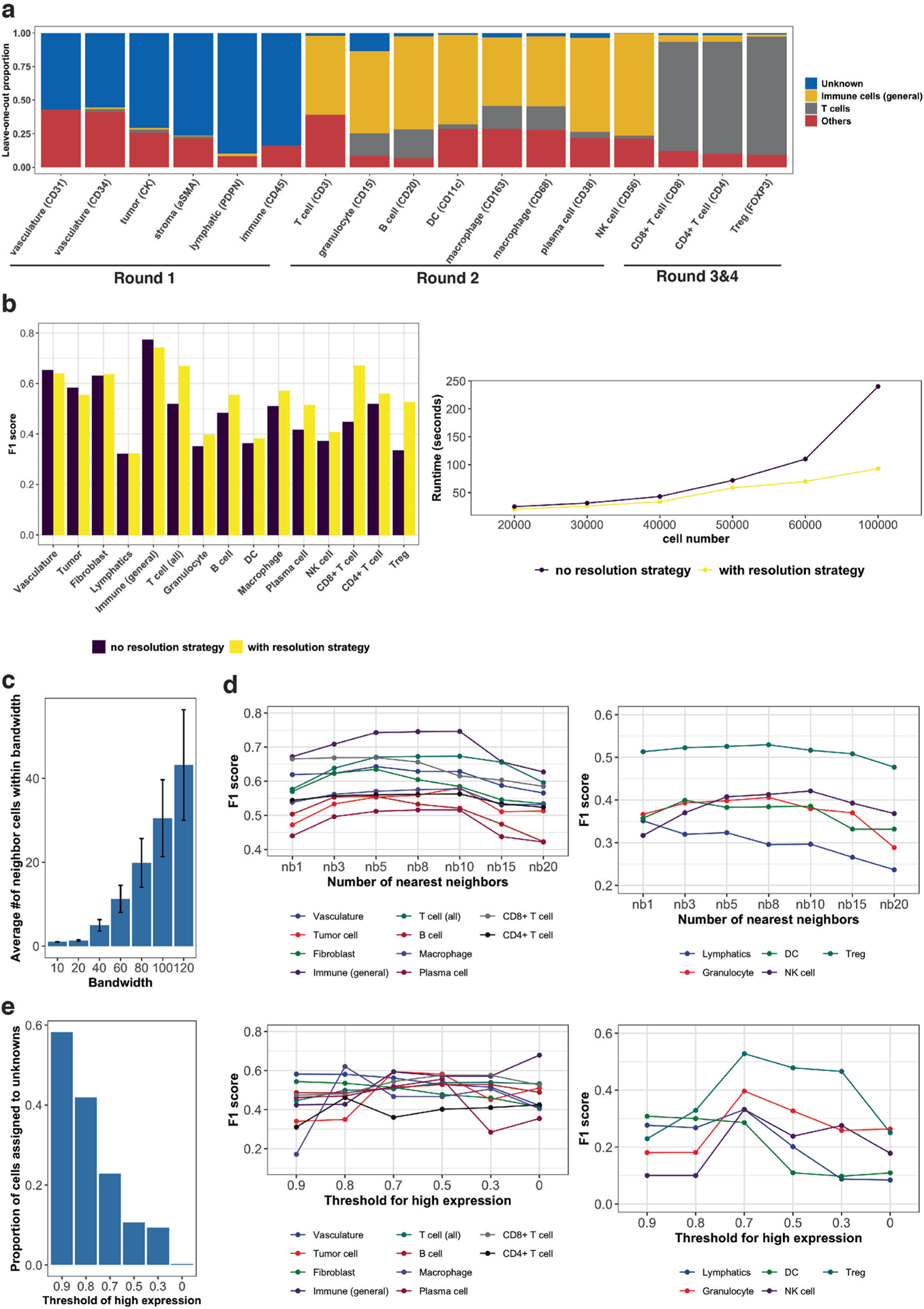
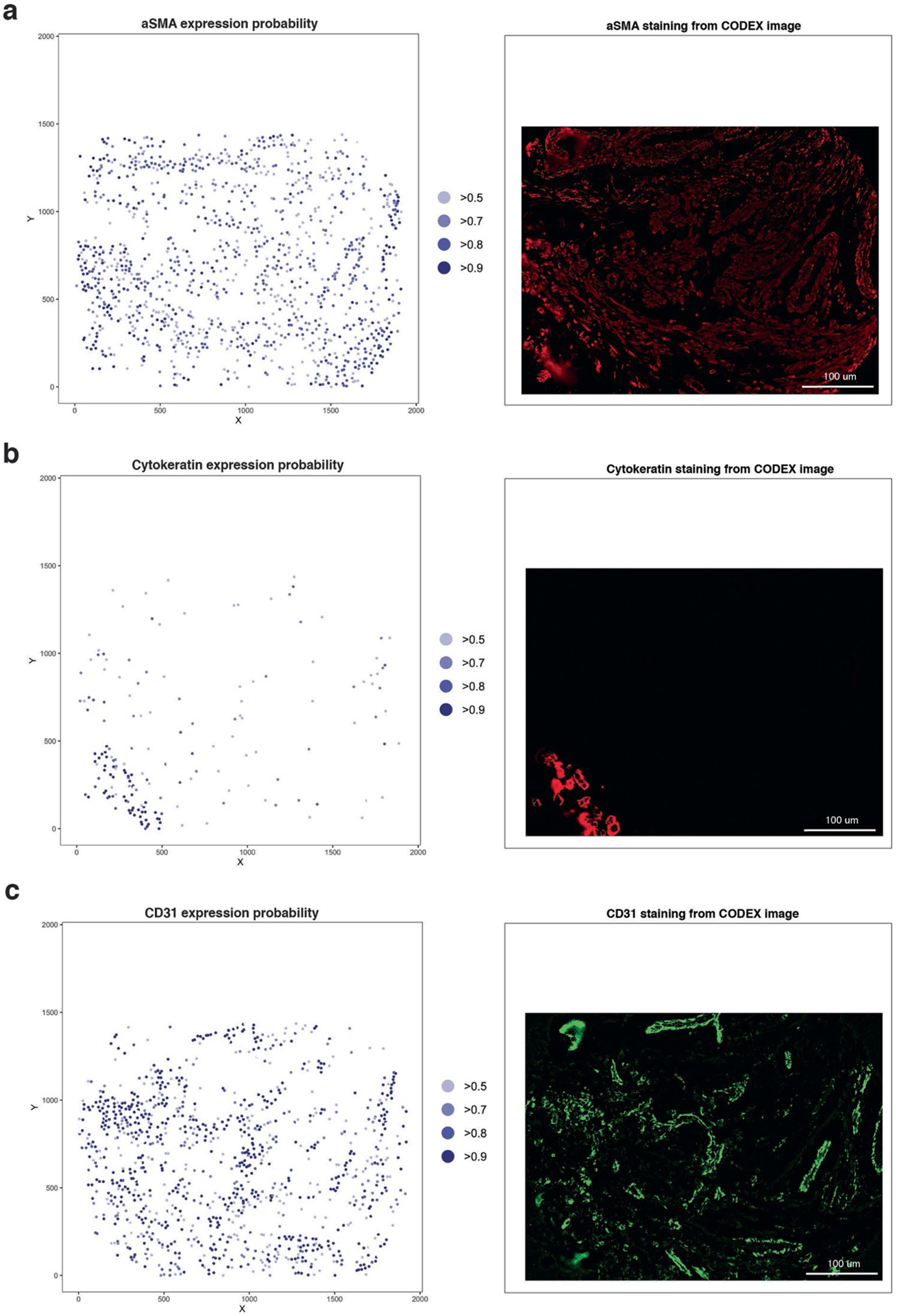
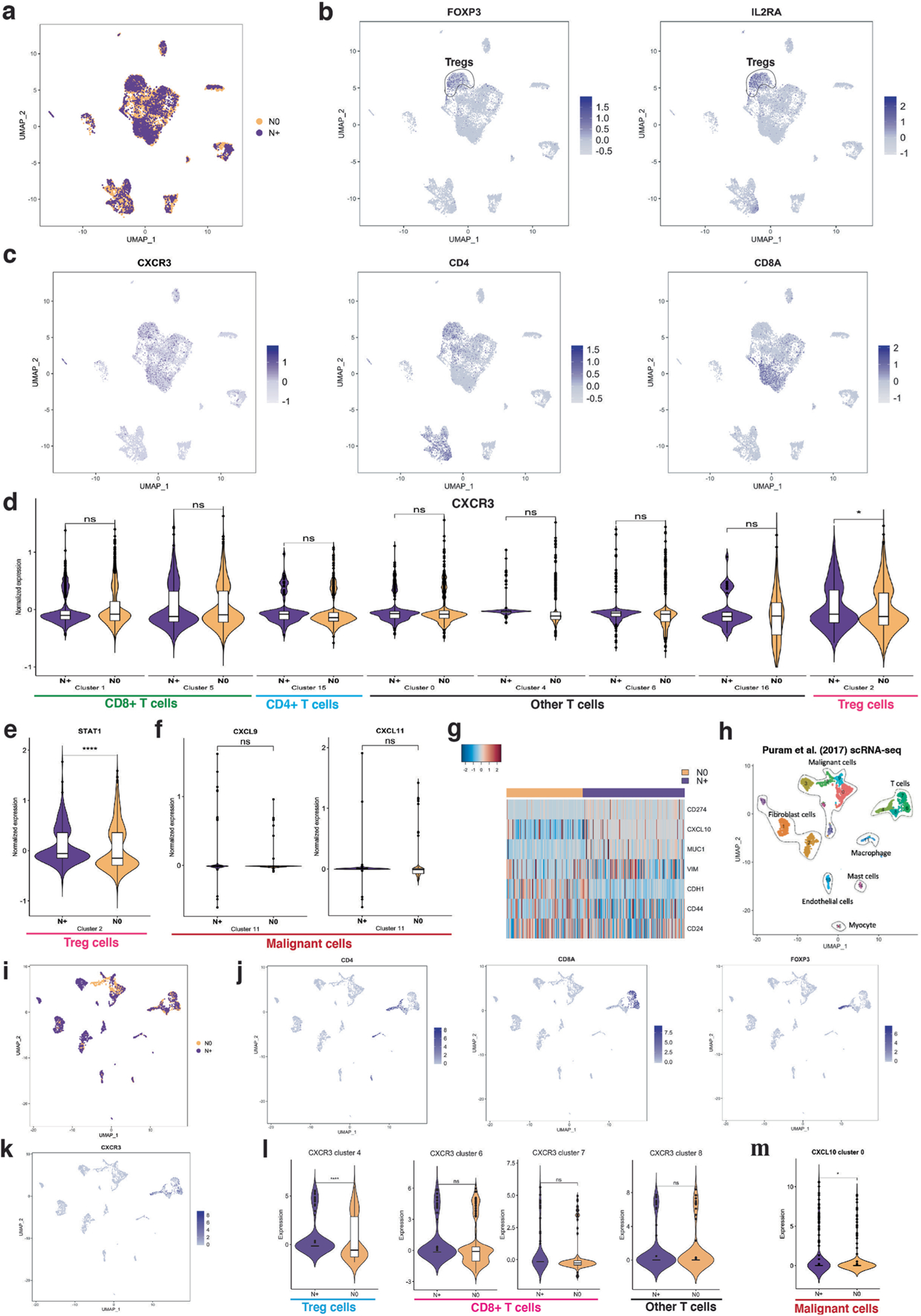
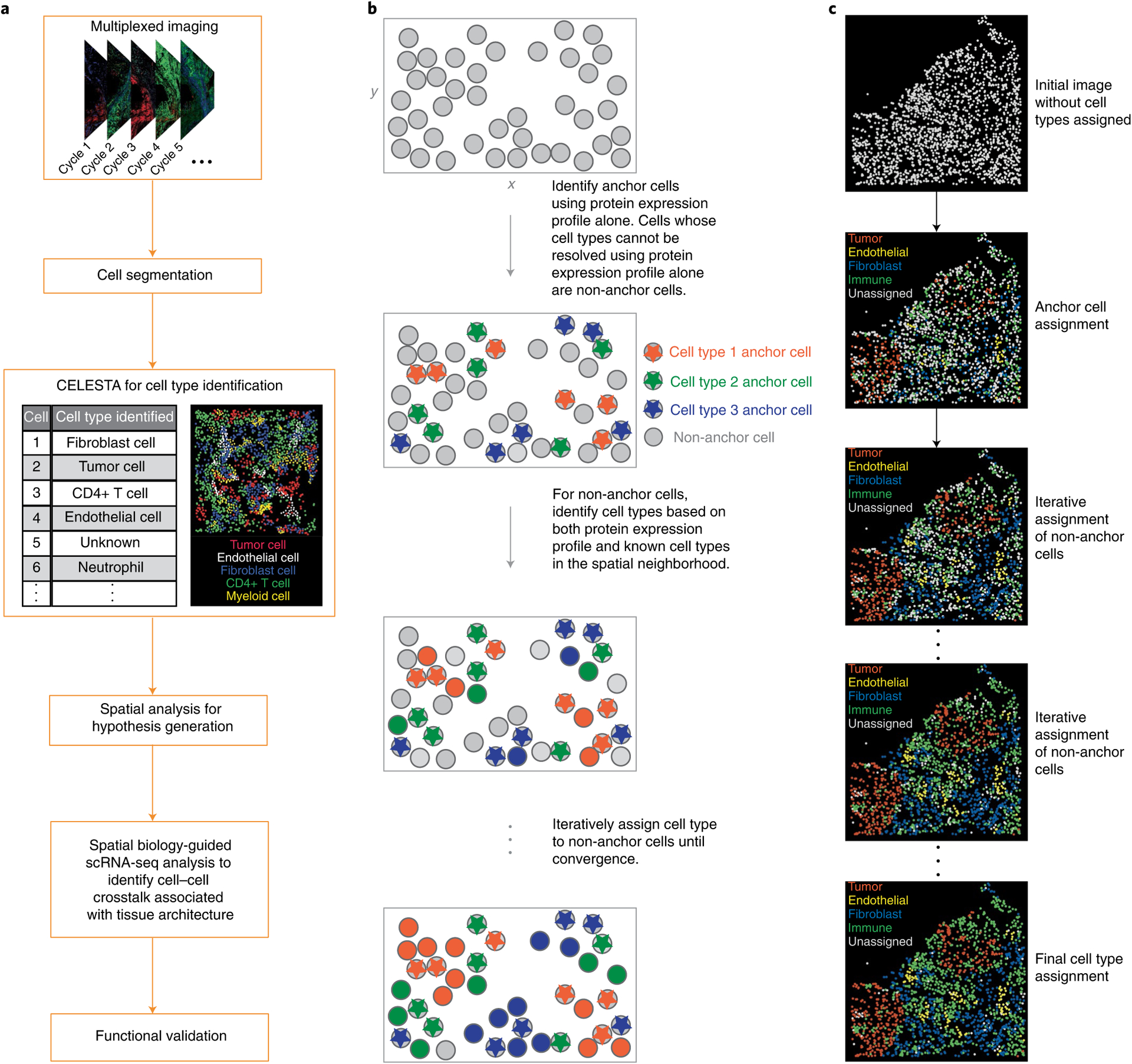
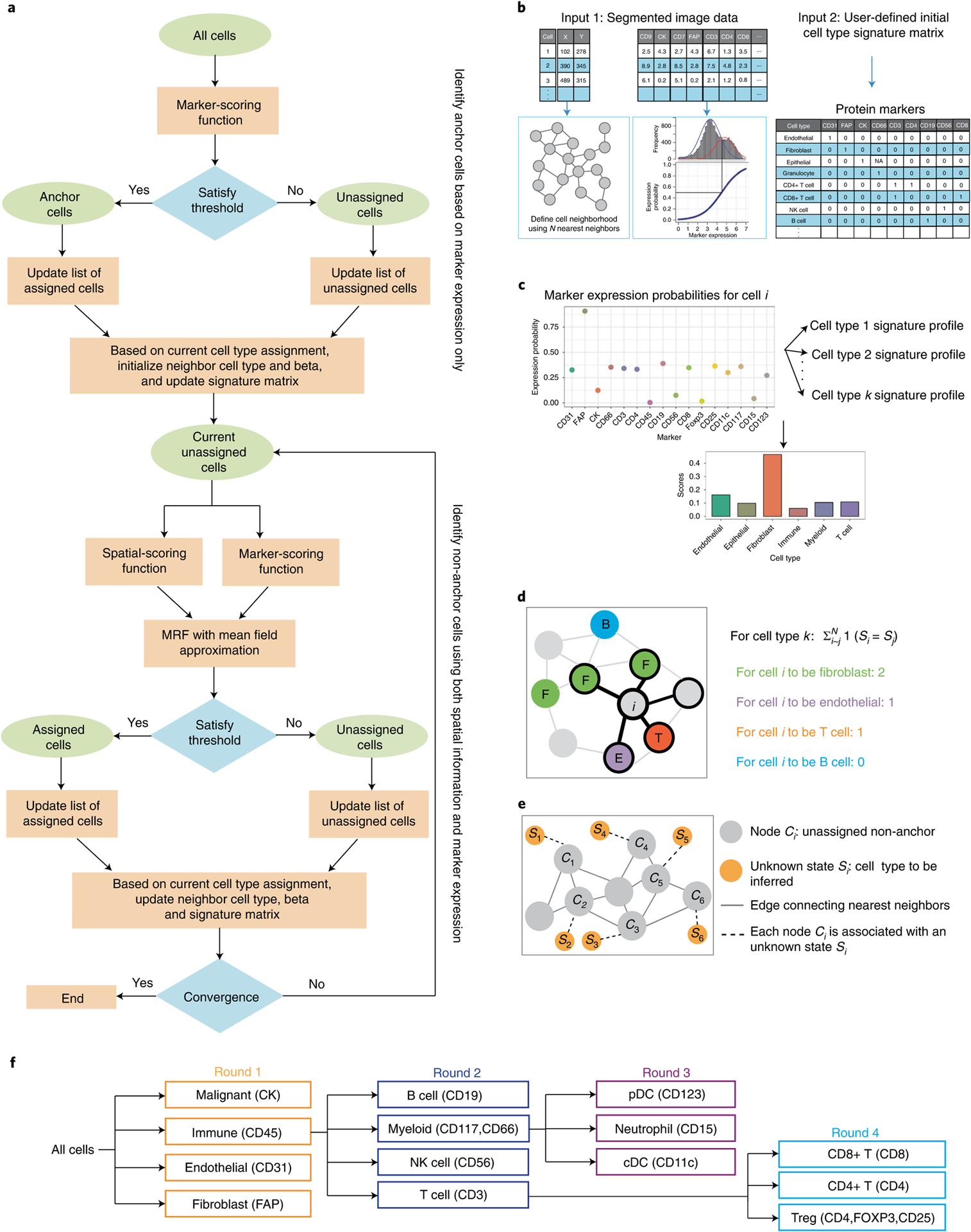
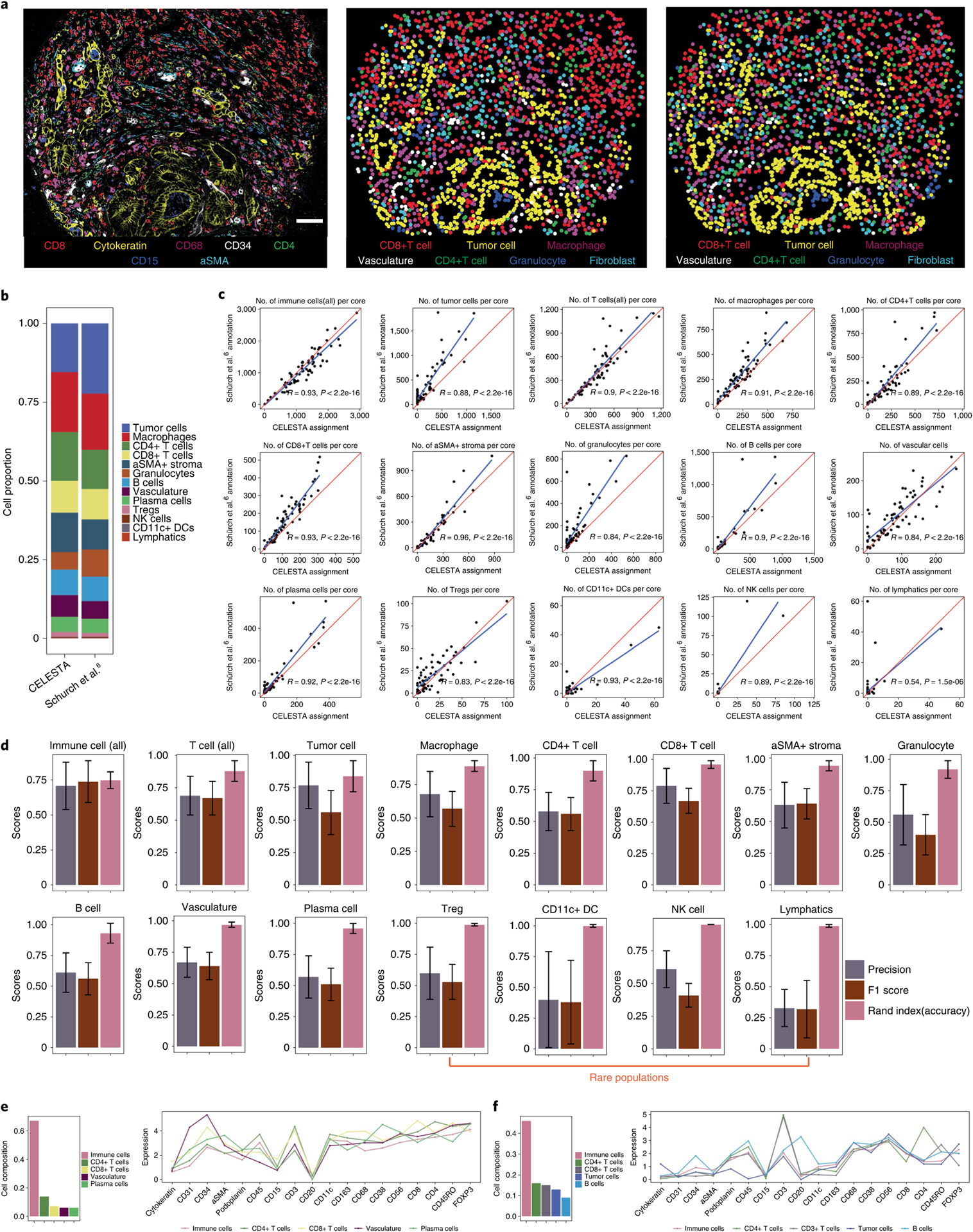
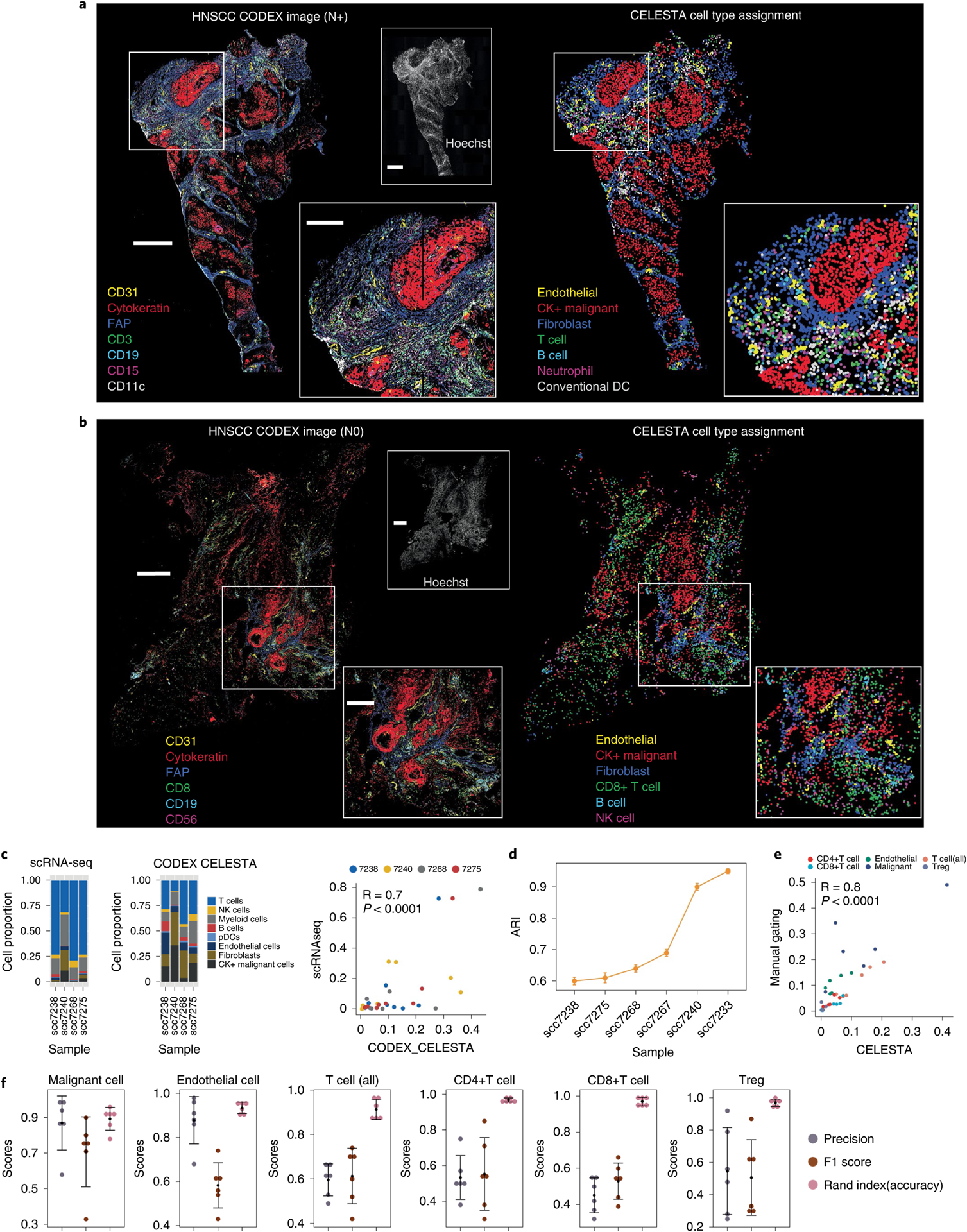
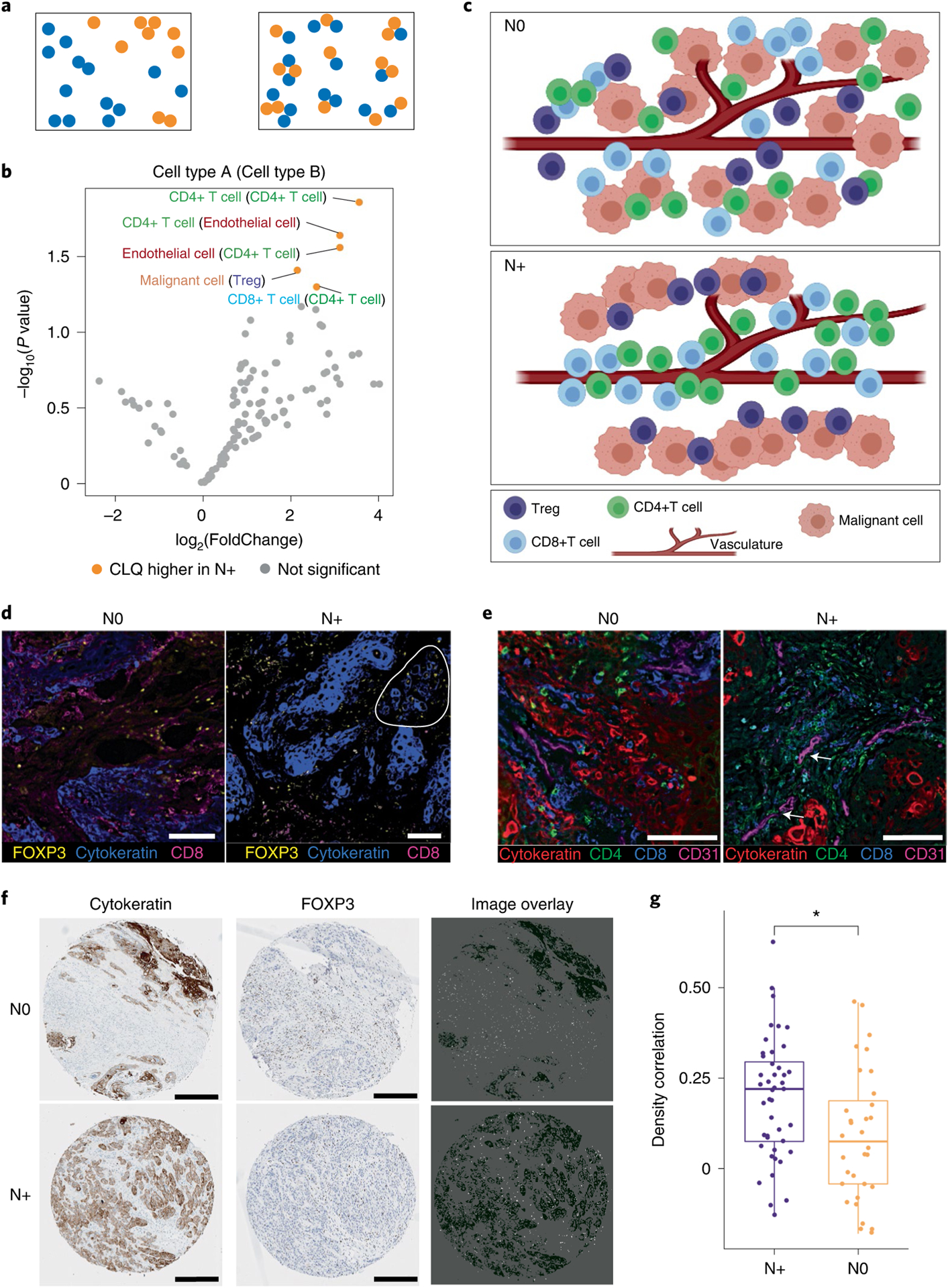
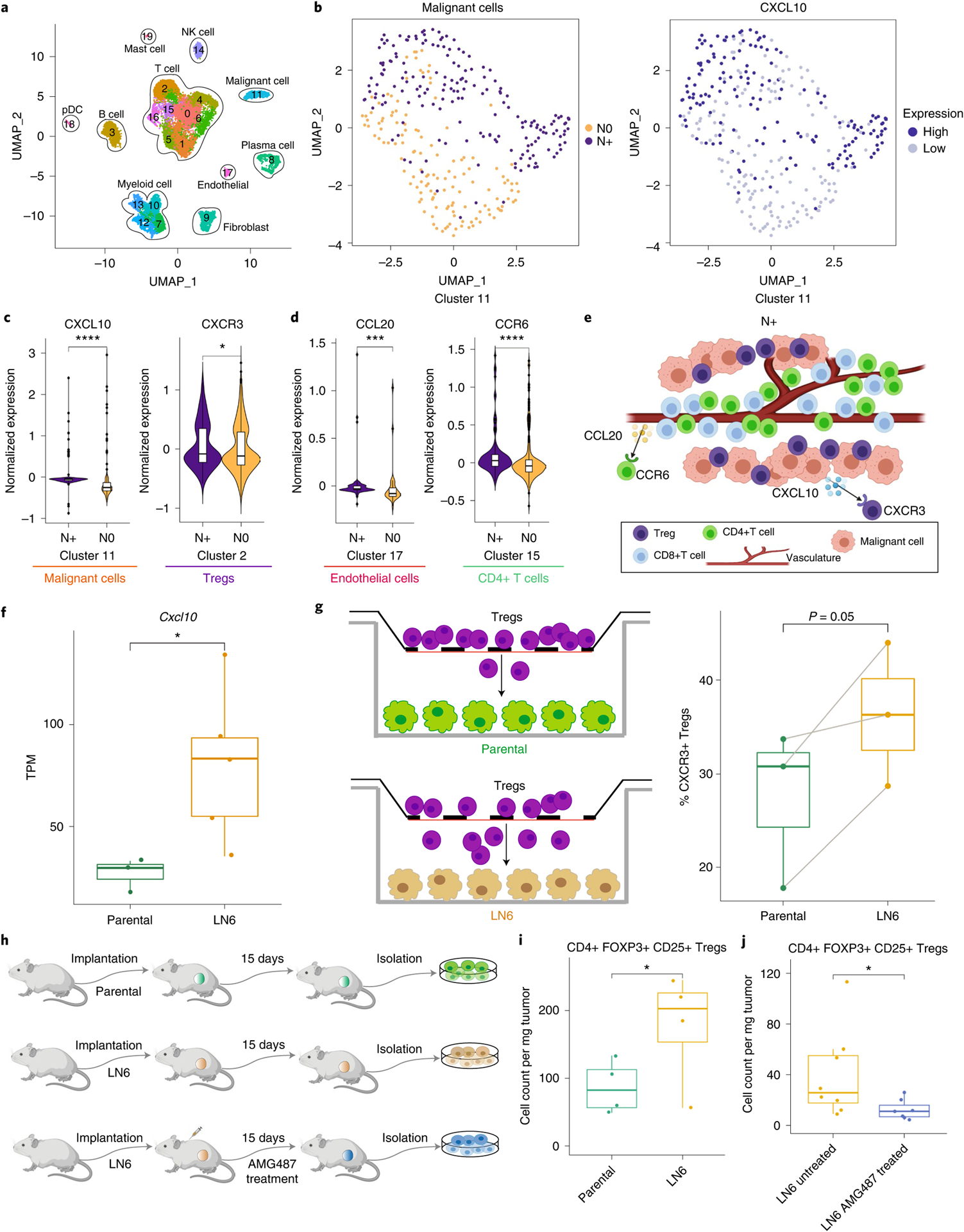
Advances in multiplexed in situ imaging are revealing important insights in spatial biology. However, cell type identification remains a major challenge in imaging analysis, with most existing methods involving substantial manual assessment and subjective decisions for thousands of cells. We developed an unsupervised machine learning algorithm, CELESTA, which identifies the cell type of each cell, individually, using the cell's marker expression profile and, when needed, its spatial information. We demonstrate the performance of CELESTA on multiplexed immunofluorescence images of colorectal cancer and head and neck squamous cell carcinoma (HNSCC). Using the cell types identified by CELESTA, we identify tissue architecture associated with lymph node metastasis in HNSCC, and validate our findings in an independent cohort. By coupling our spatial analysis with single-cell RNA-sequencing data on proximal sections of the same specimens, we identify cell-cell crosstalk associated with lymph node metastasis, demonstrating the power of CELESTA to facilitate identification of clinically relevant interactions.
© 2022. The Author(s), under exclusive licence to Springer Nature America, Inc.
Figures
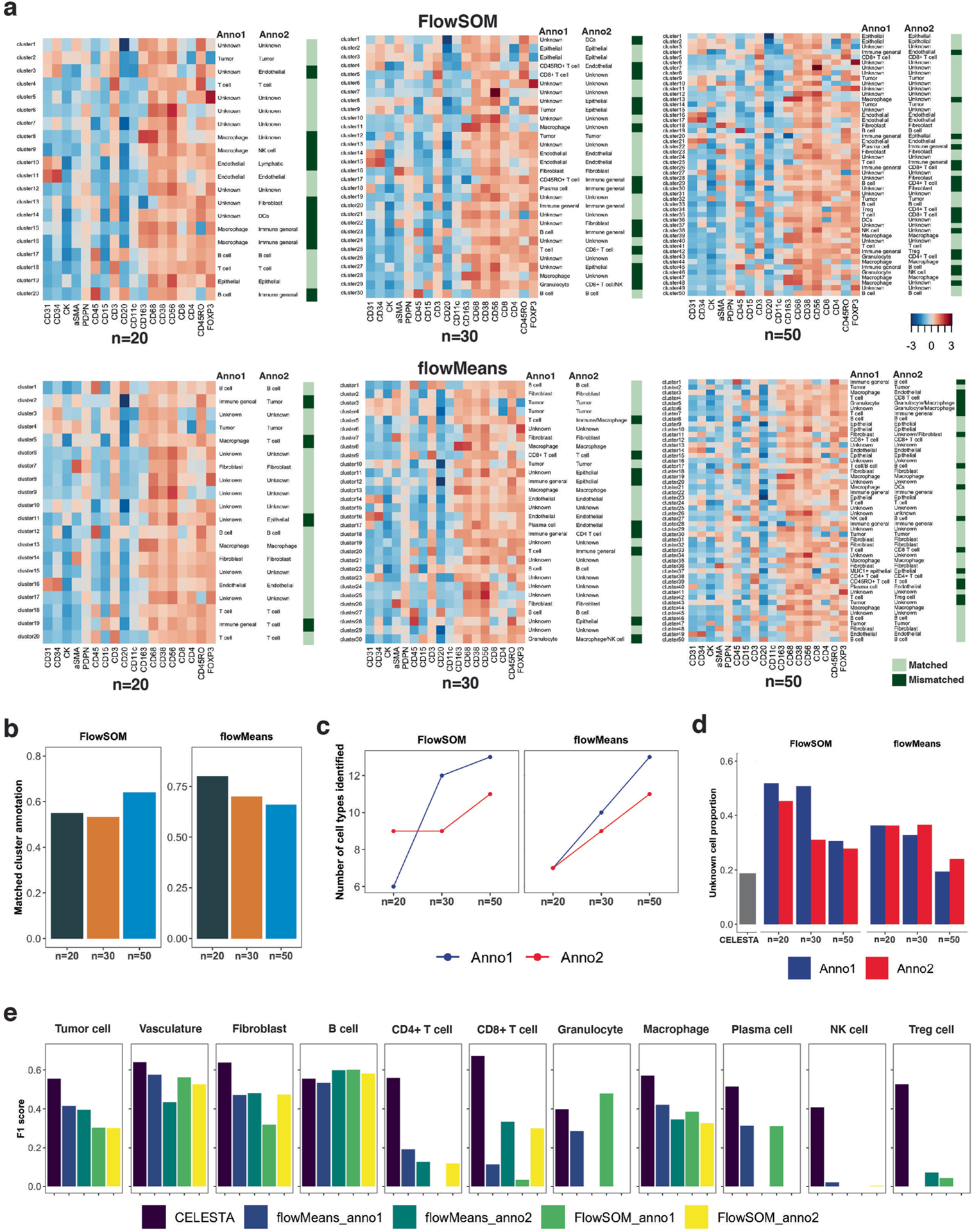
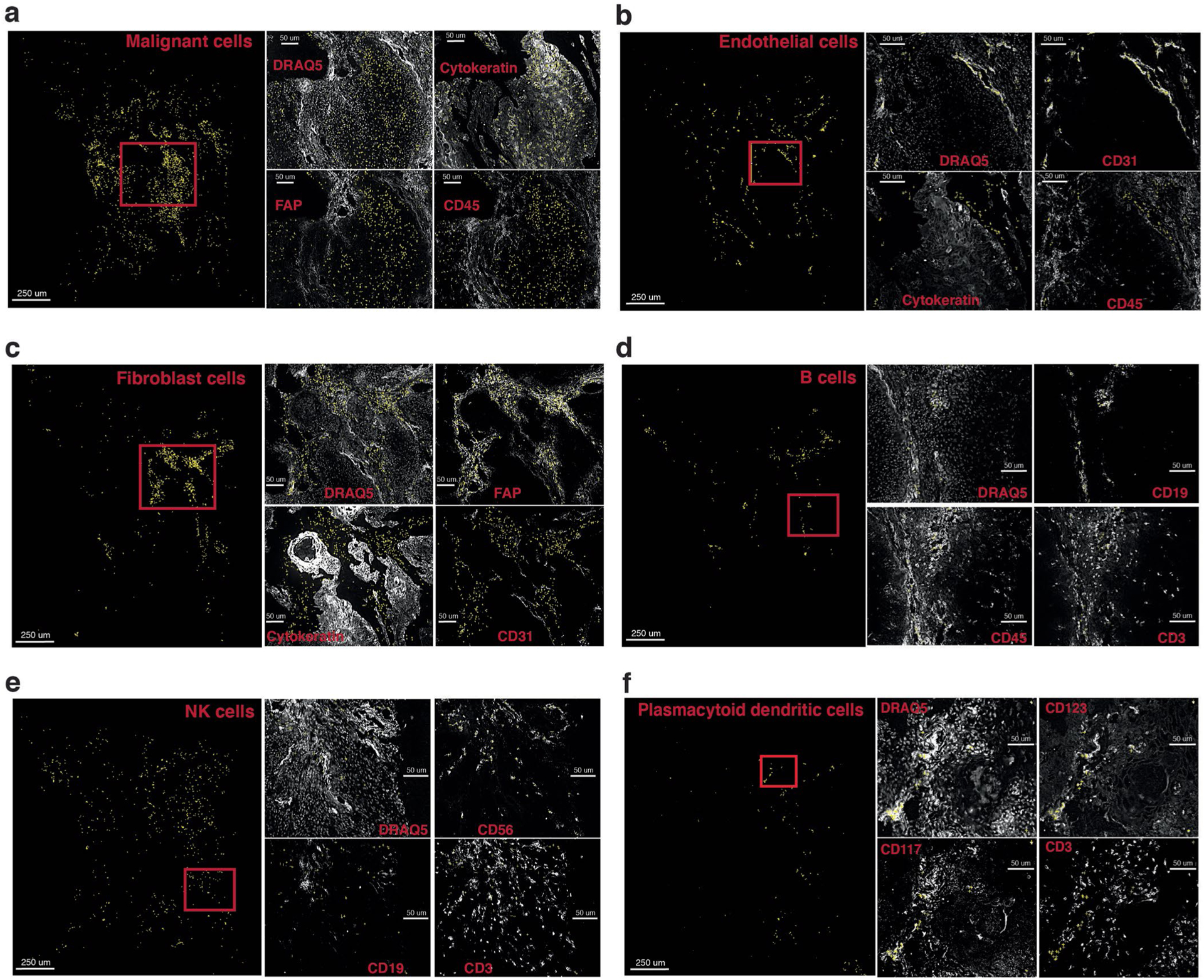
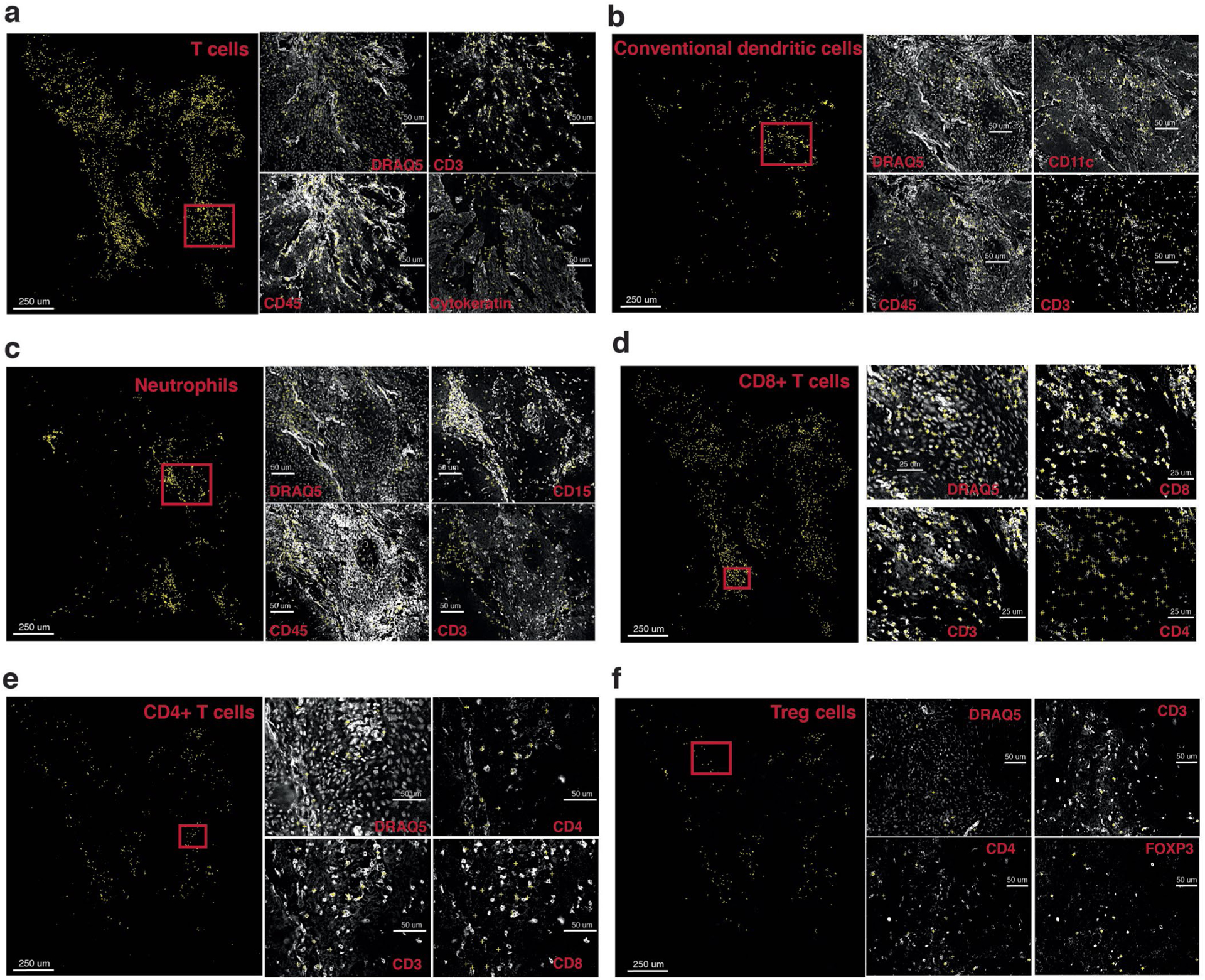
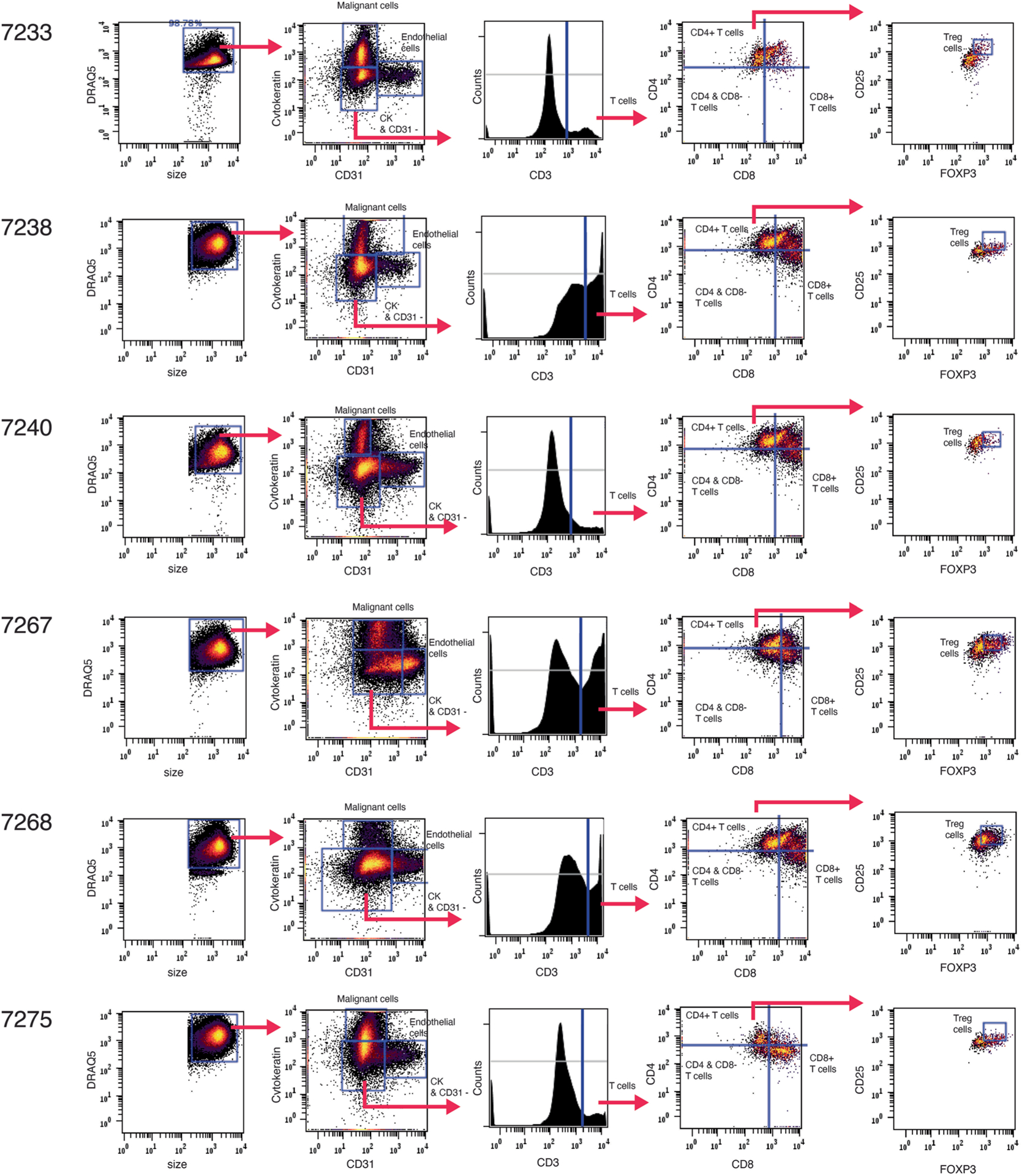
References
-
- Stack EC, Wang C, Roman KA & Hoyt CC Multiplexed immunohistochemistry, imaging, and quantitation: a review, with an assessment of tyramide signal amplification, multispectral imaging and multiplex analysis. Methods 70, 46–58 (2014). - PubMed
-
- Ptacek J et al. Multiplexed ion beam imaging (MIBI) for characterization of the tumor microenvironment across tumor types. Lab. Invest 100, 1111–1123 (2020). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
