

Screening and Whole Genome Sequencing of SARS-CoV-2 Circulating During the First Three Waves of the COVID-19 Pandemic in Libreville and the Haut-Ogooué Province in Gabon
- PMID: 35655849
- PMCID: PMC9152426
- DOI: 10.3389/fmed.2022.877391
Screening and Whole Genome Sequencing of SARS-CoV-2 Circulating During the First Three Waves of the COVID-19 Pandemic in Libreville and the Haut-Ogooué Province in Gabon
Abstract
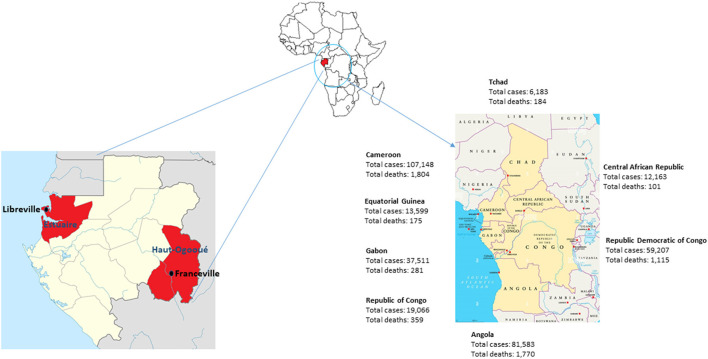
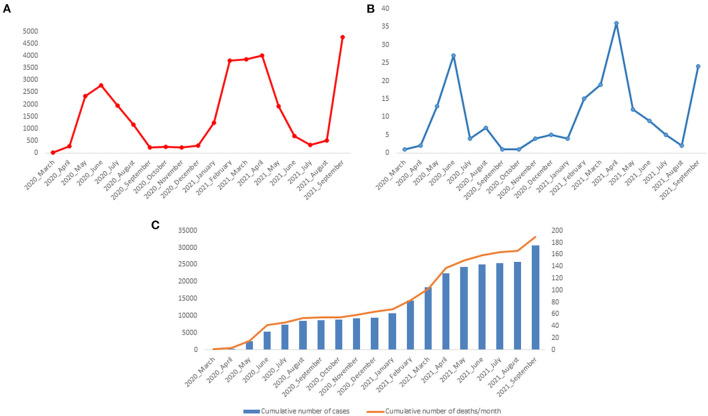
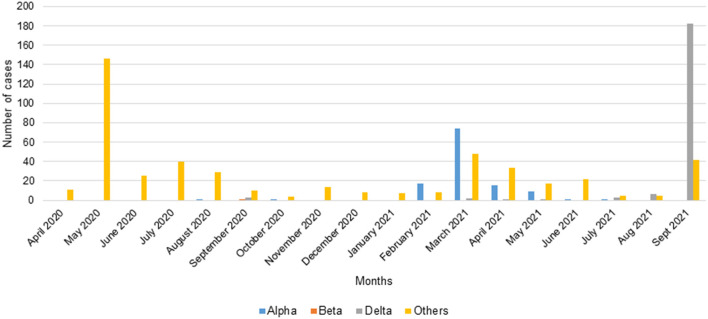
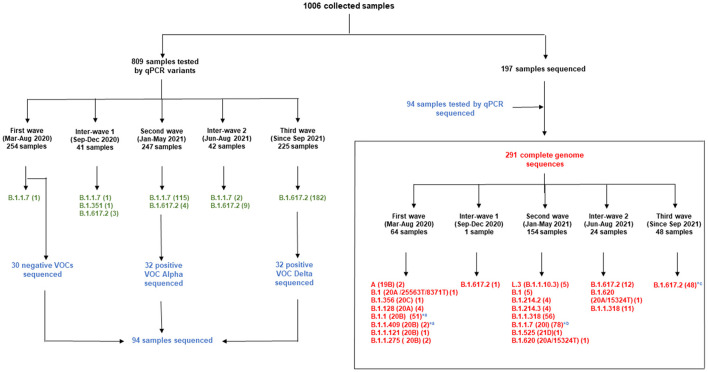
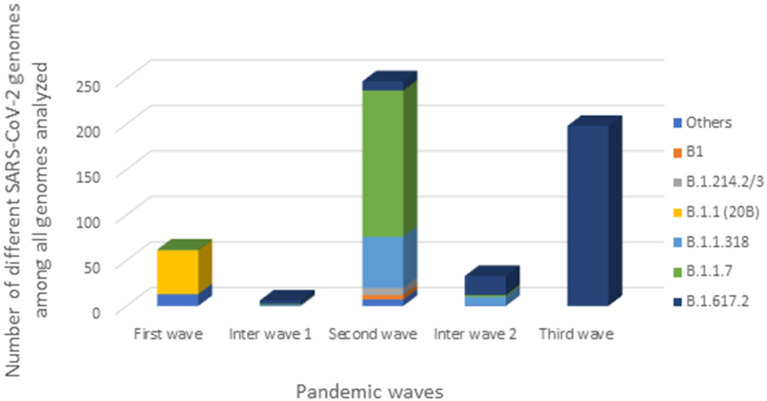
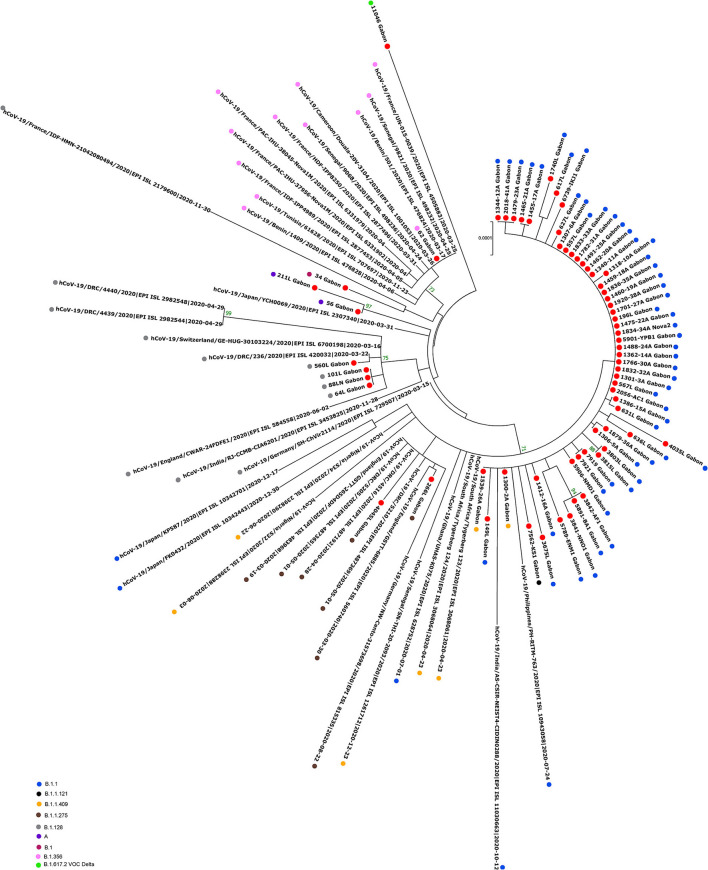
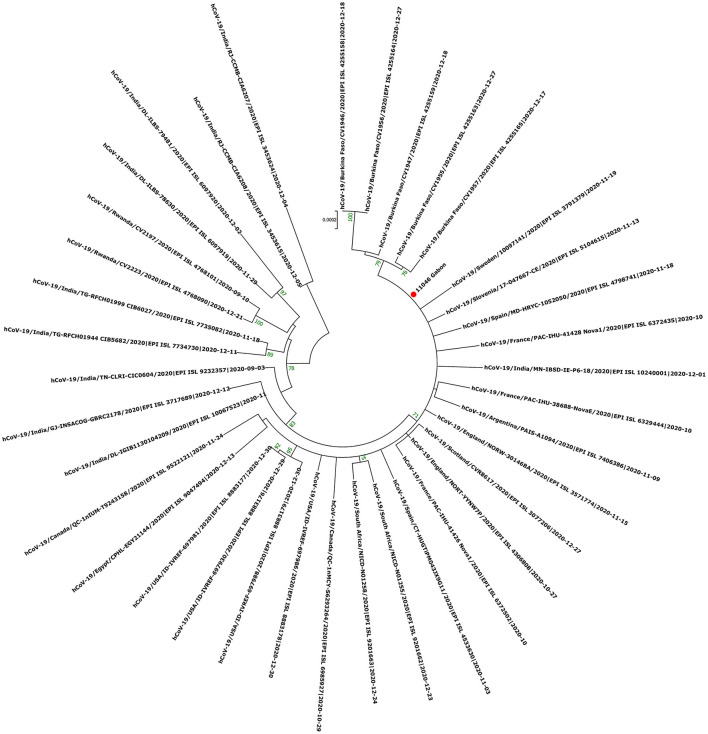
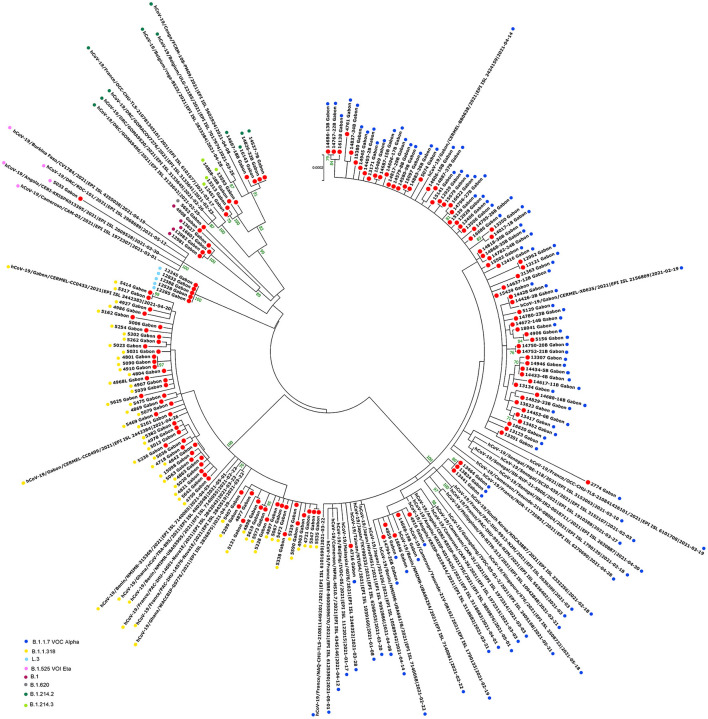
Since the onset of the COVID-19 pandemic, the SARS-CoV-2 viral dynamics in Africa have been less documented than on other continents. In Gabon, a Central African country, a total number of 37,511 cases of COVID-19 and 281 deaths have been reported as of December 8, 2021. After the first COVID-19 case was reported on March 12, 2020, in the capital Libreville, the country experienced two successive waves. The first one, occurred in March 2020 to August 2020, and the second one in January 2021 to May 2021. The third wave began in September 2021 and ended in November 2021. In order to reduce the data gap regarding the dynamics of SARS-CoV-2 in Central Africa, we performed a retrospective genotyping study using 1,006 samples collected from COVID-19 patients in Gabon from 2020 to 2021. Using SARS-CoV-2 variant screening by Real-Time Quantitative Reverse Transcription PCR (qRT-PCR) and whole genome sequencing (WGS), we genotyped 809 SARS-CoV-2 samples through qRT-PCR and identified to generated 291 new genomes. It allowed us to describe specific mutations and changes in the SARS-CoV-2 variants in Gabon. The qRT-PCR screening of 809 positive samples from March 2020 to September 2021 showed that 119 SARS-CoV-2 samples (14.7%) were classified as VOC Alpha (Pangolin lineage B.1.1.7), one (0.1%) was a VOC Beta (B.1.351), and 198 (24.5 %) were VOC Delta (B.1.617.2), while 491 samples (60.7%) remained negative for the variants sought. The B1.1 variant was predominant during the first wave while the VOC Alpha dominated the second wave. The B1.617.2 Delta variant is currently the dominant variant of the third wave. Similarly, the analysis of the 291 genome sequences indicated that the dominant variant during the first wave was lineage B.1.1, while the dominant variants of the second wave were lineages B.1.1.7 (50.6%) and B.1.1.318 (36.4%). The third wave started with the circulation of the Delta variant (B.1.617). Finally, we compared these results to the SARS-CoV-2 sequences reported in other African, European, American and Asian countries. Sequences of Gabonese SARS-CoV-2 strains presented the highest similarities with those of France, Belgium and neighboring countries of Central Africa, as well as West Africa.
Keywords: Gabon; Libreville and Haut-Ogooué; SARS-CoV-2; variant; whole genome sequencing.
Copyright © 2022 Lekana-Douki, N'dilimabaka, Levasseur, Colson, Andeko, Zong Minko, Banga Mve-Ella, Fournier, Devaux, Ondo, Akombi, Yacka Mouele Bolo, Ngonga Dikongo, Diané, Mabika Mabika, Mathouet, Dzembo, Atiga, Mouity Matoumba, Ndjangangoye, Bréchard, Bedotto-Buffet, Mangombi Pambou, Kandet Yattara, Mbongo Nkama, Mintsa Ndong, Adegnika, Raoult, Fenollar and Lekana-Douki.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
Similar articles
-
COVID-19 in 16 West African Countries: An Assessment of the Epidemiology and Genetic Diversity of SARS-CoV-2 after Four Epidemic Waves.Am J Trop Med Hyg. 2023 Aug 28;109(4):861-873. doi: 10.4269/ajtmh.22-0469. Print 2023 Oct 4. Am J Trop Med Hyg. 2023. PMID: 37640294 Free PMC article. Review.
-
The dynamics of circulating SARS-CoV-2 lineages in Bogor and surrounding areas reflect variant shifting during the first and second waves of COVID-19 in Indonesia.PeerJ. 2022 Mar 22;10:e13132. doi: 10.7717/peerj.13132. eCollection 2022. PeerJ. 2022. PMID: 35341058 Free PMC article.
-
Spatiotemporal prevalence of COVID-19 and SARS-CoV-2 variants in Africa.Front Public Health. 2025 Feb 20;13:1526727. doi: 10.3389/fpubh.2025.1526727. eCollection 2025. Front Public Health. 2025. PMID: 40051513 Free PMC article.
-
SARS-CoV-2 Variants by Whole-Genome Sequencing in a University Hospital in Bangkok: First to Third COVID-19 Waves.Pathogens. 2023 Apr 21;12(4):626. doi: 10.3390/pathogens12040626. Pathogens. 2023. PMID: 37111512 Free PMC article.
-
Genomic evolution of the SARS-CoV-2 Variants of Concern: COVID-19 pandemic waves in India.EXCLI J. 2023 Jun 1;22:451-465. doi: 10.17179/excli2023-6098. eCollection 2023. EXCLI J. 2023. PMID: 37534220 Free PMC article. Review.
Cited by
-
COVID-19 in 16 West African Countries: An Assessment of the Epidemiology and Genetic Diversity of SARS-CoV-2 after Four Epidemic Waves.Am J Trop Med Hyg. 2023 Aug 28;109(4):861-873. doi: 10.4269/ajtmh.22-0469. Print 2023 Oct 4. Am J Trop Med Hyg. 2023. PMID: 37640294 Free PMC article. Review.
-
Tracing Emergence of SARS-CoV-2 Variants: Insights from Comprehensive Assessment Using Reverse Transcription Polymerase Chain Reaction and Whole Genome Sequencing.Microorganisms. 2025 Jan 31;13(2):311. doi: 10.3390/microorganisms13020311. Microorganisms. 2025. PMID: 40005678 Free PMC article.
-
COVIDSeq as Laboratory Developed Test (LDT) for Diagnosis of SARS-CoV-2 Variants of Concern (VOC).Arch Clin Biomed Res. 2022;6(6):954-970. doi: 10.26502/acbr.50170309. Epub 2022 Nov 28. Arch Clin Biomed Res. 2022. PMID: 36588916 Free PMC article.
-
Unravelling Antigenic Cross-Reactions toward the World of Coronaviruses: Extent of the Stability of Shared Epitopes and SARS-CoV-2 Anti-Spike Cross-Neutralizing Antibodies.Pathogens. 2023 May 13;12(5):713. doi: 10.3390/pathogens12050713. Pathogens. 2023. PMID: 37242383 Free PMC article. Review.
-
Molecular epidemiology of SARS-CoV-2 in Northern South Africa: wastewater surveillance from January 2021 to May 2022.Front Public Health. 2023 Dec 19;11:1309869. doi: 10.3389/fpubh.2023.1309869. eCollection 2023. Front Public Health. 2023. PMID: 38174083 Free PMC article.
References
LinkOut - more resources
Full Text Sources
Miscellaneous