

Histone Deacetylase 3: A Potential Therapeutic Target for Atherosclerosis
- PMID: 35656103
- PMCID: PMC9116907
- DOI: 10.14336/AD.2021.1116
Histone Deacetylase 3: A Potential Therapeutic Target for Atherosclerosis
Abstract
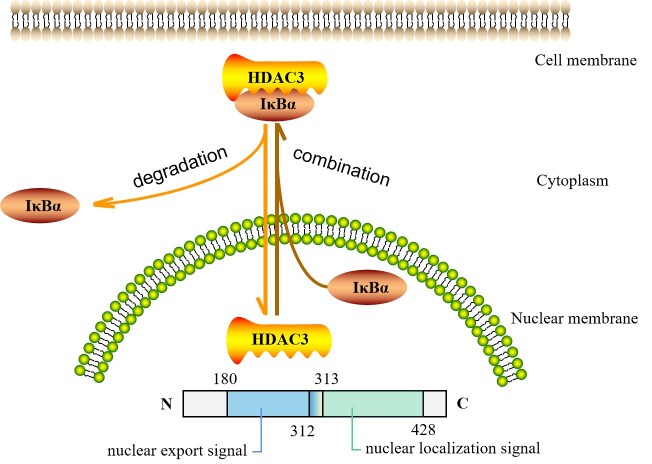
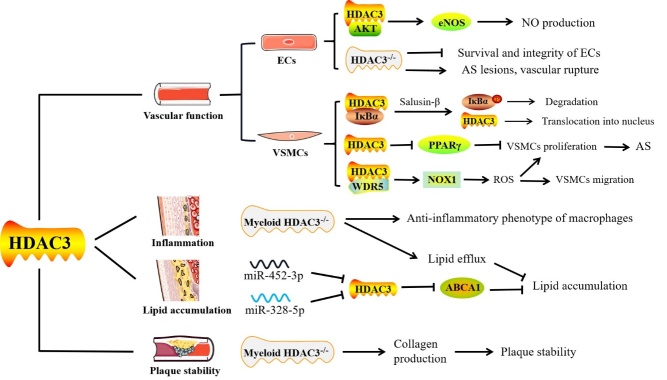
Atherosclerosis, the pathological basis of most cardiovascular disease, is characterized by plaque formation in the intima. Secondary lesions include intraplaque hemorrhage, plaque rupture, and local thrombosis. Vascular endothelial function impairment and smooth muscle cell migration lead to vascular dysfunction, which is conducive to the formation of macrophage-derived foam cells and aggravates inflammatory response and lipid accumulation that cause atherosclerosis. Histone deacetylase (HDAC) is an epigenetic modifying enzyme closely related to chromatin structure and gene transcriptional regulation. Emerging studies have demonstrated that the Class I member HDAC3 of the HDAC super family has cell-specific functions in atherosclerosis, including 1) maintenance of endothelial integrity and functions, 2) regulation of vascular smooth muscle cell proliferation and migration, 3) modulation of macrophage phenotype, and 4) influence on foam cell formation. Although several studies have shown that HDAC3 may be a promising therapeutic target, only a few HDAC3-selective inhibitors have been thoroughly researched and reported. Here, we specifically summarize the impact of HDAC3 and its inhibitors on vascular function, inflammation, lipid accumulation, and plaque stability in the development of atherosclerosis with the hopes of opening up new opportunities for the treatment of cardiovascular diseases.
Keywords: HDAC3; HDAC3 inhibitors; acetylation; atherosclerosis; cardiovascular diseases.
Copyright: © 2022 Jiang et al.
Conflict of interest statement
Competing interests The authors declare that there are no disputes of interest.
Figures
References
-
- Libby P (2021). Inflammation in Atherosclerosis-No Longer a Theory. Clin chem, 67(1): 131-142. - PubMed
-
- Asare Y, Campbell-James TA, Bokov Y, Yu LL, Prestel M, El Bounkari O, et al. (2020). Histone Deacetylase 9 Activates IKK to Regulate Atherosclerotic Plaque Vulnerability. Circ Res, 127(6): 811-823. - PubMed
-
- Yang WM, Tsai SC, Wen YD, Fejer G, Seto E (2002). Functional domains of histone deacetylase-3. J Biol Chem, 277(11): 9447-54. - PubMed
Publication types
LinkOut - more resources
Full Text Sources