

The Liver-α-Cell Axis in Health and in Disease
- PMID: 35657688
- PMCID: PMC9862287
- DOI: 10.2337/dbi22-0004
The Liver-α-Cell Axis in Health and in Disease
Abstract
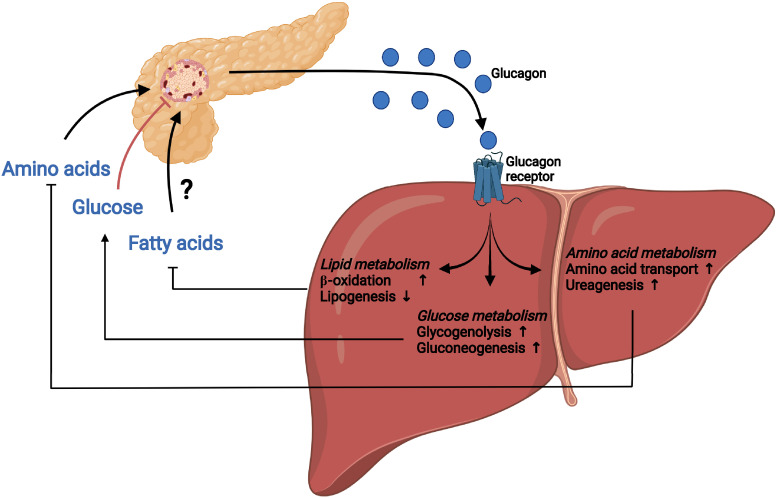
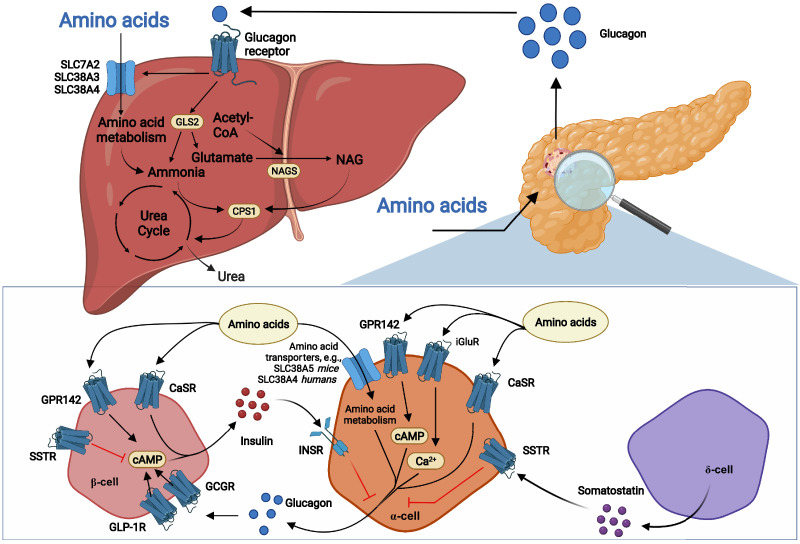
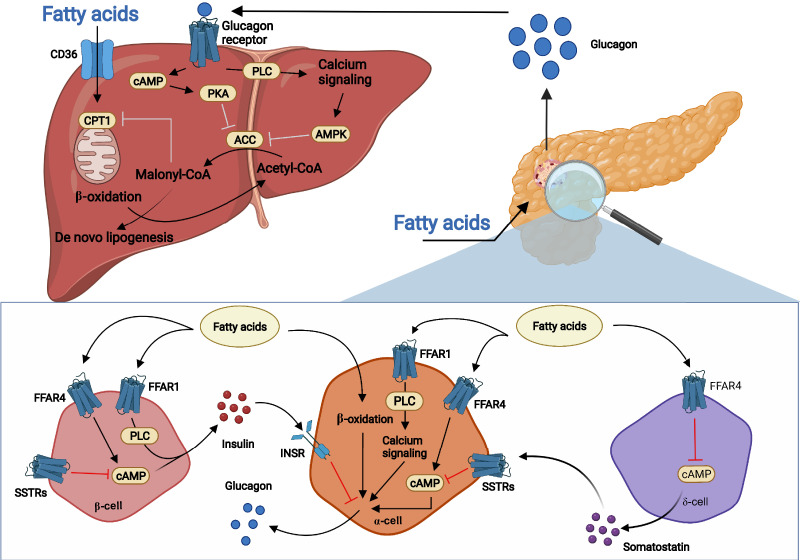
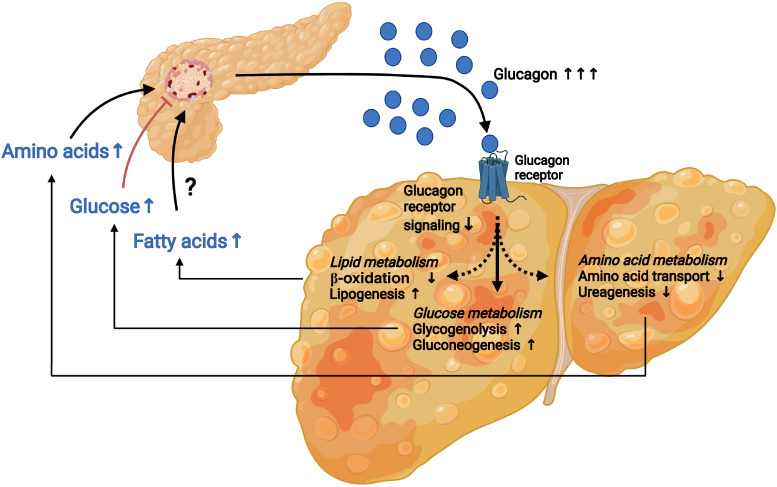
Glucagon and insulin are the main regulators of blood glucose. While the actions of insulin are extensively mapped, less is known about glucagon. Besides glucagon's role in glucose homeostasis, there are additional links between the pancreatic α-cells and the hepatocytes, often collectively referred to as the liver-α-cell axis, that may be of importance for health and disease. Thus, glucagon receptor antagonism (pharmacological or genetic), which disrupts the liver-α-cell axis, results not only in lower fasting glucose but also in reduced amino acid turnover and dyslipidemia. Here, we review the actions of glucagon on glucose homeostasis, amino acid catabolism, and lipid metabolism in the context of the liver-α-cell axis. The concept of glucagon resistance is also discussed, and we argue that the various elements of the liver-α-cell axis may be differentially affected in metabolic diseases such as diabetes, obesity, and nonalcoholic fatty liver disease (NAFLD). This conceptual rethinking of glucagon biology may explain why patients with type 2 diabetes have hyperglucagonemia and how NAFLD disrupts the liver-α-cell axis, compromising the normal glucagon-mediated enhancement of substrate-induced amino acid turnover and possibly fatty acid β-oxidation. In contrast to amino acid catabolism, glucagon-induced glucose production may not be affected by NAFLD, explaining the diabetogenic effect of NAFLD-associated hyperglucagonemia. Consideration of the liver-α-cell axis is essential to understanding the complex pathophysiology underlying diabetes and other metabolic diseases.
© 2022 by the American Diabetes Association.
Figures
References
-
- Müller TD, Finan B, Clemmensen C, DiMarchi RD, Tschöp MH. The new biology and pharmacology of glucagon. Physiol Rev 2017;97:721–766 - PubMed
-
- Wewer Albrechtsen NJ, Kuhre RE, Pedersen J, Knop FK, Holst JJ. The biology of glucagon and the consequences of hyperglucagonemia. Biomarkers Med 2016;10:1141–1151 - PubMed
-
- Scheen AJ, Paquot N, Lefèbvre PJ. Investigational glucagon receptor antagonists in phase I and II clinical trials for diabetes. Expert Opin Investig Drugs 2017;26:1373–1389 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
