

Targeting mitochondrial DNA polymerase gamma for selective inhibition of MLH1 deficient colon cancer growth
- PMID: 35657956
- PMCID: PMC9165880
- DOI: 10.1371/journal.pone.0268391
Targeting mitochondrial DNA polymerase gamma for selective inhibition of MLH1 deficient colon cancer growth
Abstract
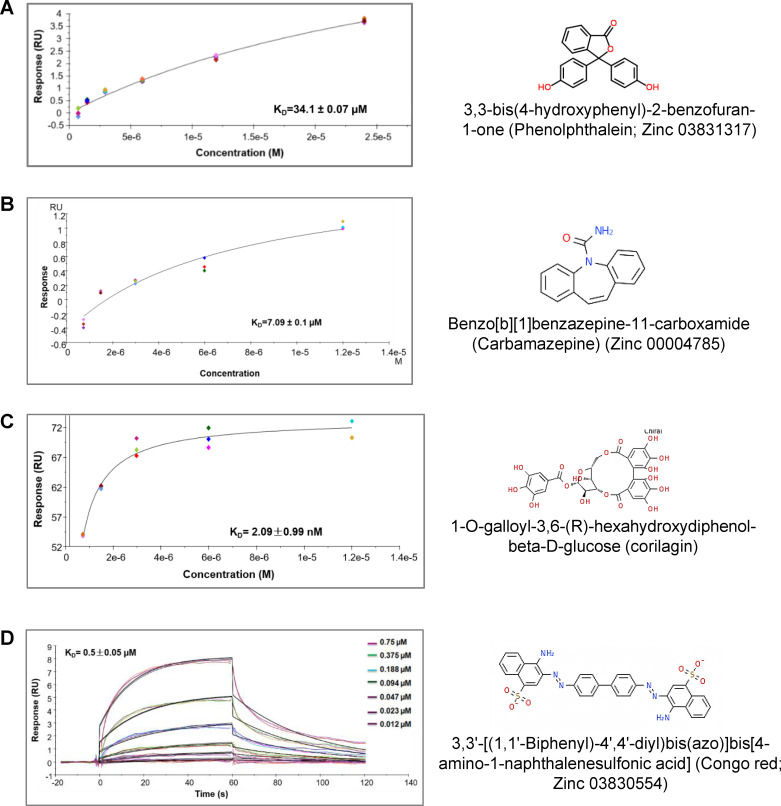
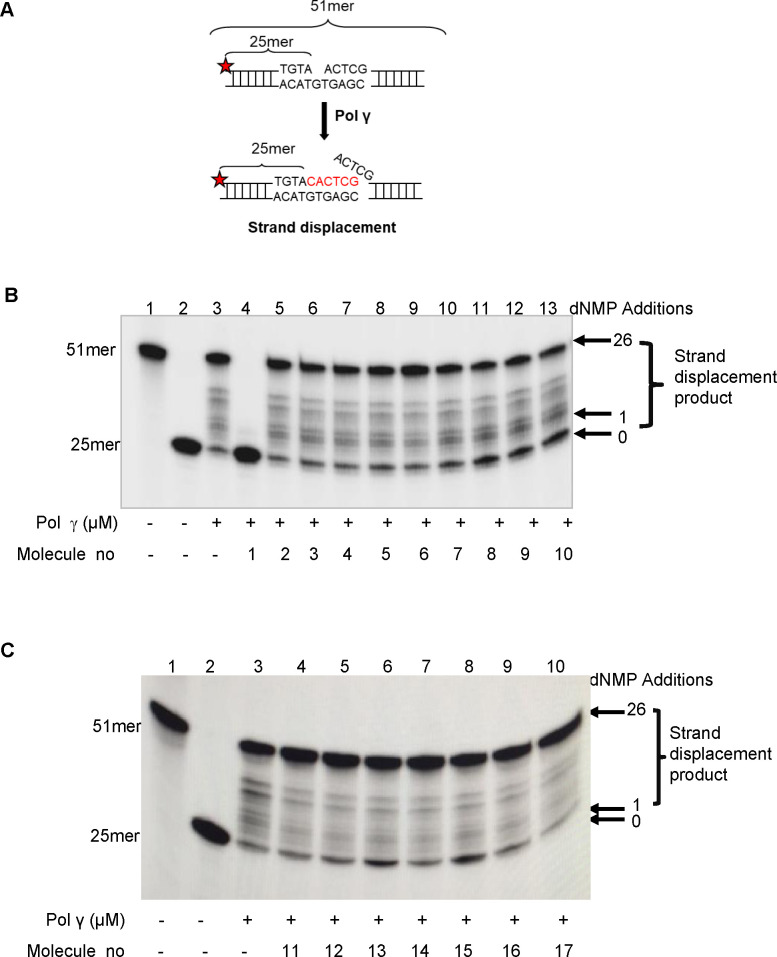
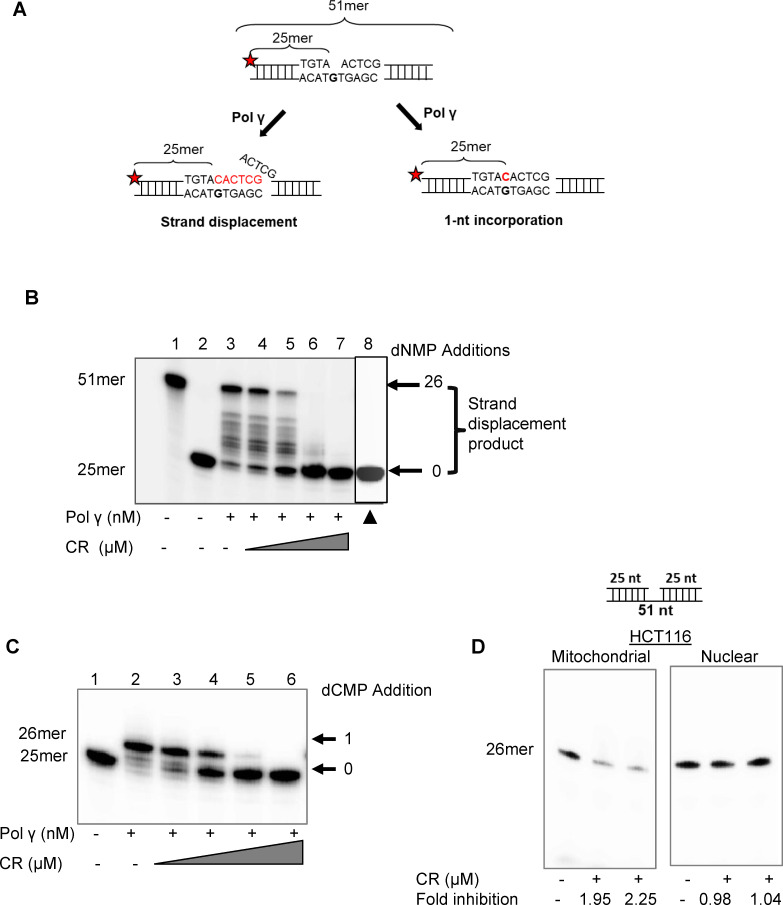
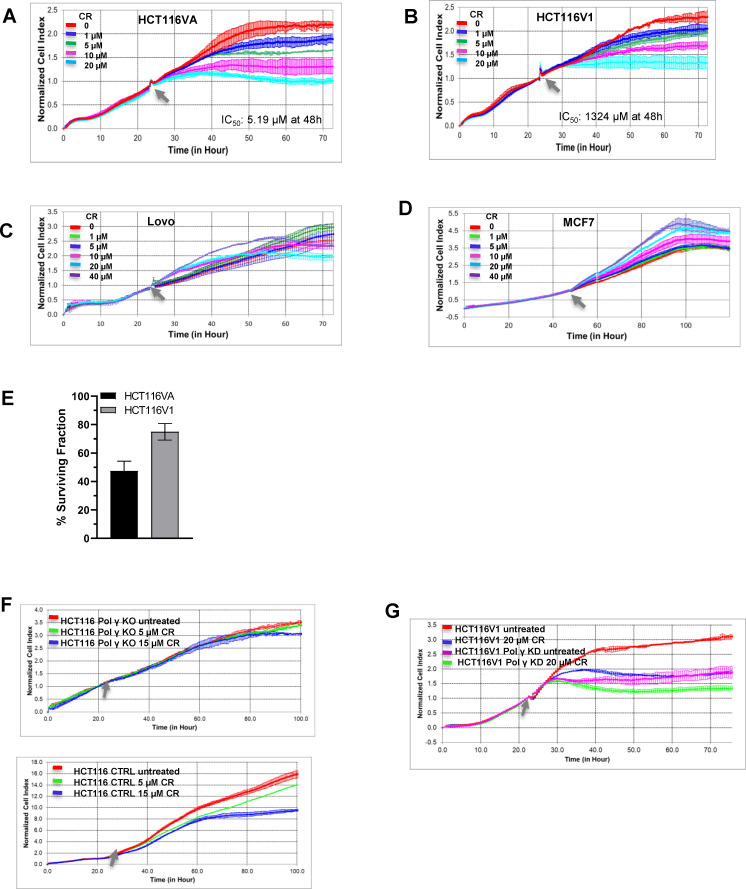
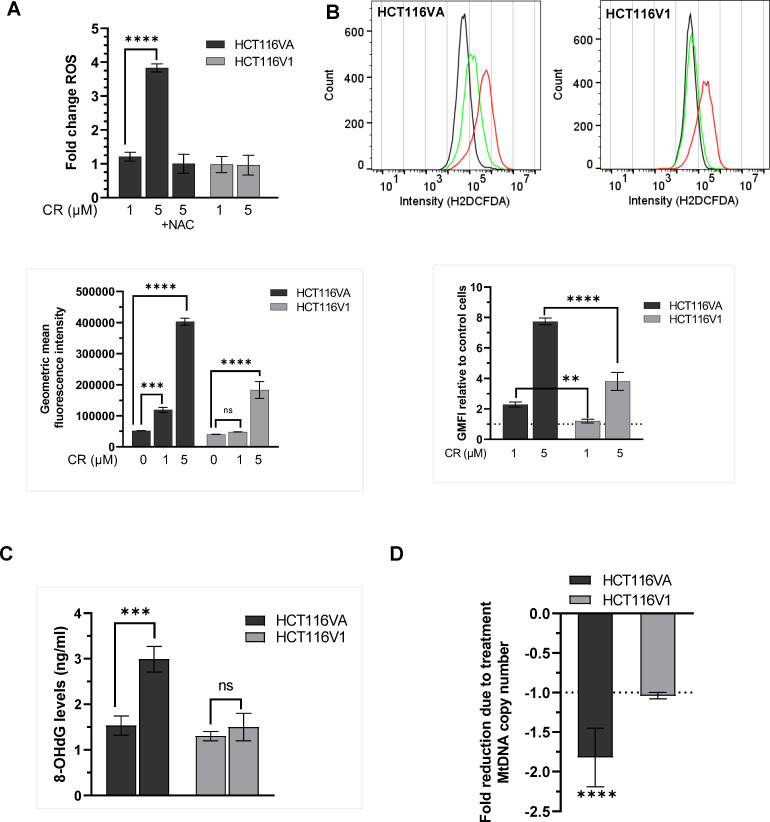
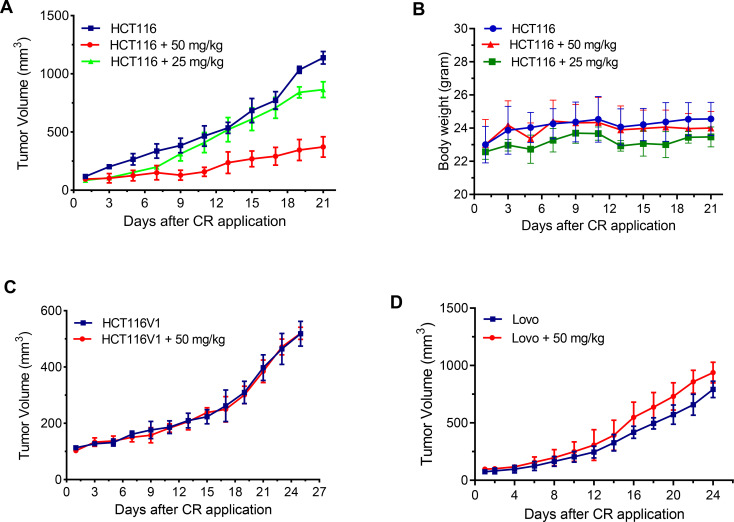
Synthetic lethality in DNA repair pathways is an important strategy for the selective treatment of cancer cells without harming healthy cells and developing cancer-specific drugs. The synthetic lethal interaction between the mismatch repair (MMR) protein, MutL homolog 1 (MLH1), and the mitochondrial base excision repair protein, DNA polymerase γ (Pol γ) was used in this study for the selective treatment of MLH1 deficient cancers. Germline mutations in the MLH1 gene and aberrant MLH1 promoter methylation result in an increased risk of developing many cancers, including nonpolyposis colorectal and endometrial cancers. Because the inhibition of Pol γ in MLH1 deficient cancer cells provides the synthetic lethal selectivity, we conducted a comprehensive small molecule screening from various databases and chemical drug library molecules for novel Pol γ inhibitors that selectively kill MLH1 deficient cancer cells. We characterized these Pol γ inhibitor molecules in vitro and in vivo, and identified 3,3'-[(1,1'-Biphenyl)-4',4'-diyl)bis(azo)]bis[4-amino-1-naphthalenesulfonic acid] (congo red; CR; Zinc 03830554) as a high-affinity binder to the Pol γ protein and potent inhibitor of the Pol γ strand displacement and one-nucleotide incorporation DNA synthesis activities in vitro and in vivo. CR reduced the cell proliferation of MLH1 deficient HCT116 human colon cancer cells and suppressed HCT116 xenograft tumor growth whereas it did not affect the MLH1 proficient cell proliferation and xenograft tumor growth. CR caused mitochondrial dysfunction and cell death by inhibiting Pol γ activity and oxidative mtDNA damage repair, increasing the production of reactive oxygen species and oxidative mtDNA damage in MLH1 deficient cells. This study suggests that the Pol γ inhibitor, CR may be further evaluated for the MLH1 deficient cancers' therapy.
Conflict of interest statement
MM, BuE and BaE are co-inventors on a patent application (WO2020005171). The competing interest does not alter our adherence to PLOS ONE policies on sharing data and materials.
Figures
References
-
- Martin SA, McCabe N, Mullarkey M, Cummins R, Burgess DJ, Nakabeppu Y, et al. DNA polymerases as potential therapeutic targets for cancers deficient in the DNA mismatch repair proteins MSH2 or MLH1. Cancer Cell. 2010;17(3):235–48. Epub 2010/03/17. doi: 10.1016/j.ccr.2009.12.046 ; PubMed Central PMCID: PMC2845806. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
