

scAmpi-A versatile pipeline for single-cell RNA-seq analysis from basics to clinics
- PMID: 35658001
- PMCID: PMC9200350
- DOI: 10.1371/journal.pcbi.1010097
scAmpi-A versatile pipeline for single-cell RNA-seq analysis from basics to clinics
Abstract
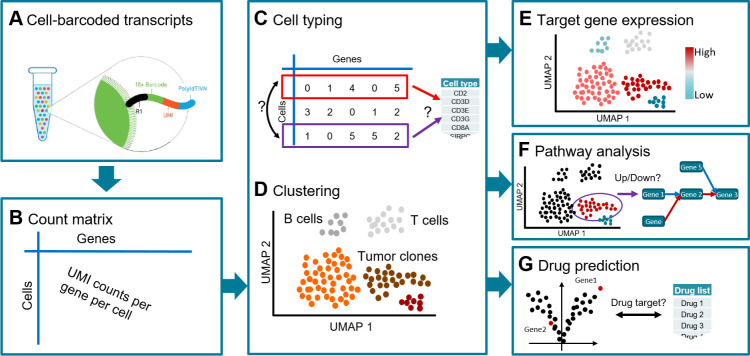
Single-cell RNA sequencing (scRNA-seq) has emerged as a powerful technique to decipher tissue composition at the single-cell level and to inform on disease mechanisms, tumor heterogeneity, and the state of the immune microenvironment. Although multiple methods for the computational analysis of scRNA-seq data exist, their application in a clinical setting demands standardized and reproducible workflows, targeted to extract, condense, and display the clinically relevant information. To this end, we designed scAmpi (Single Cell Analysis mRNA pipeline), a workflow that facilitates scRNA-seq analysis from raw read processing to informing on sample composition, clinically relevant gene and pathway alterations, and in silico identification of personalized candidate drug treatments. We demonstrate the value of this workflow for clinical decision making in a molecular tumor board as part of a clinical study.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
