

Patched 1 regulates Smoothened by controlling sterol binding to its extracellular cysteine-rich domain
- PMID: 35658032
- PMCID: PMC9166294
- DOI: 10.1126/sciadv.abm5563
Patched 1 regulates Smoothened by controlling sterol binding to its extracellular cysteine-rich domain
Abstract
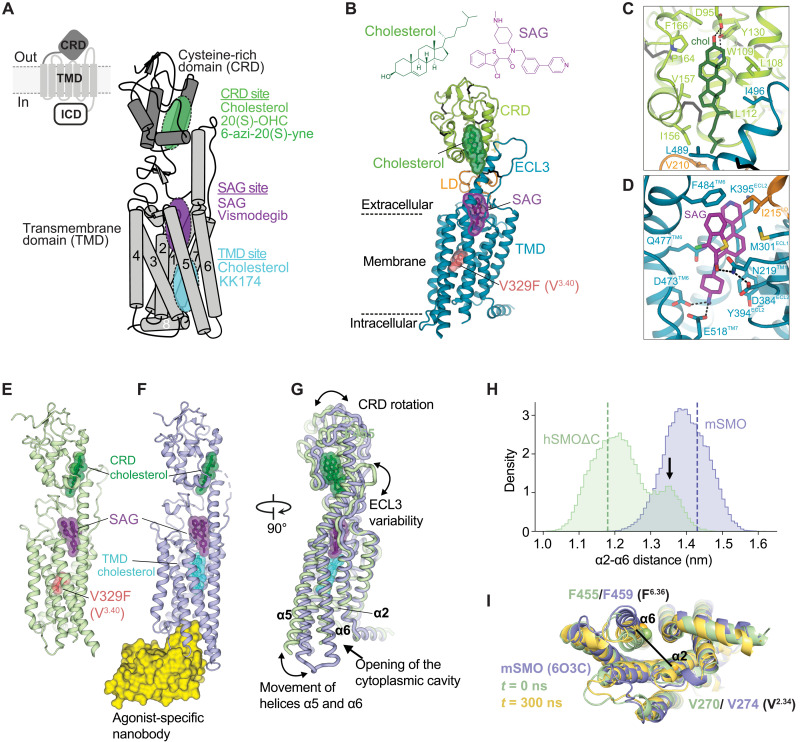
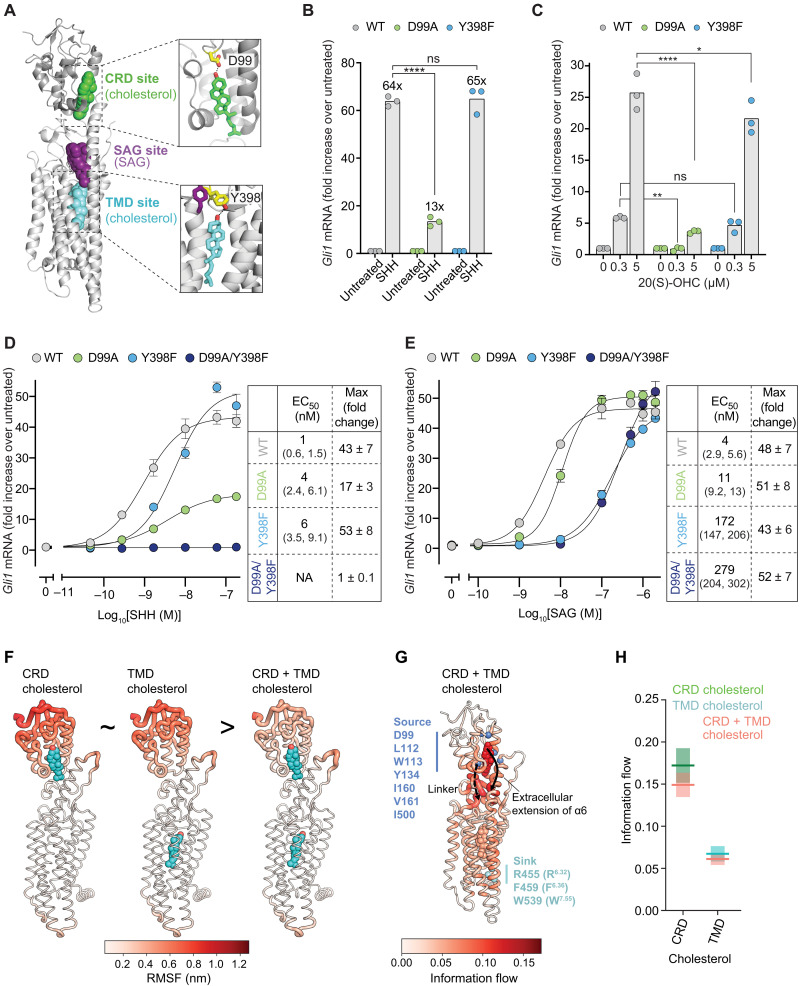
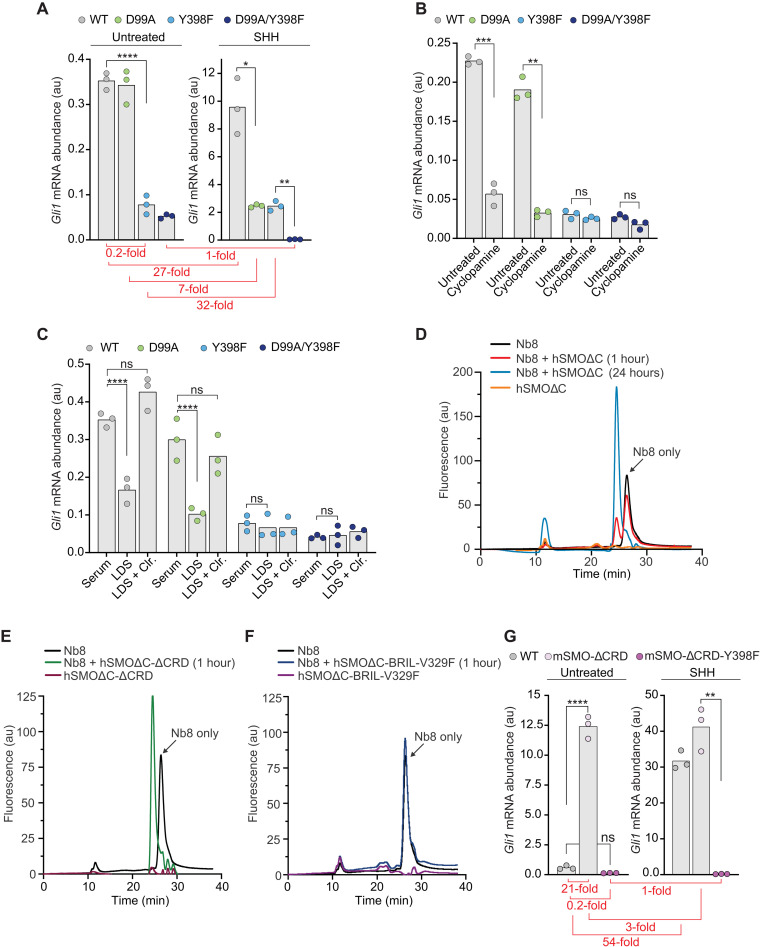
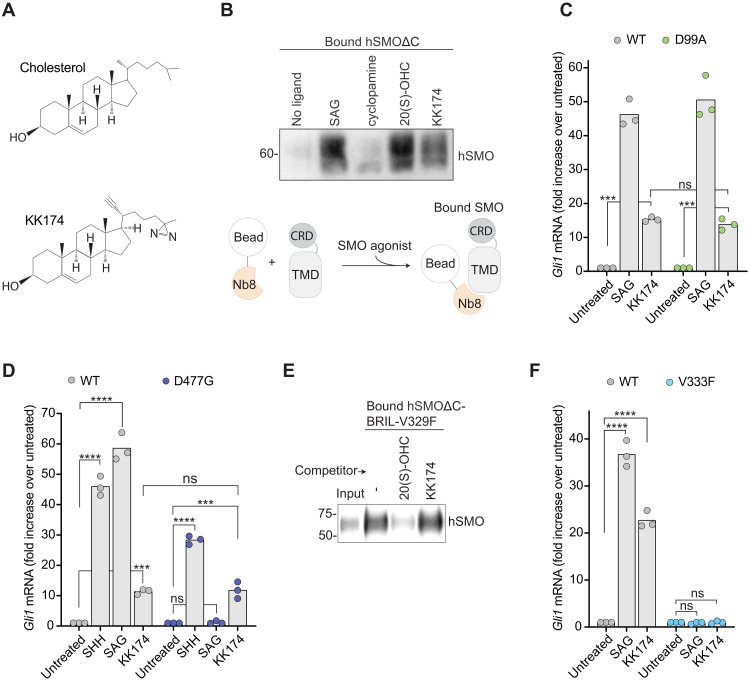
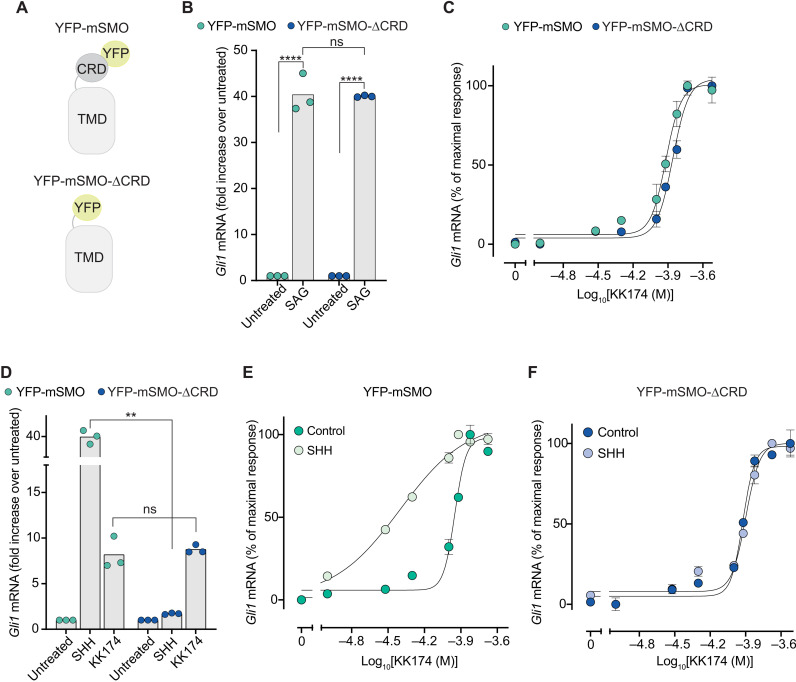
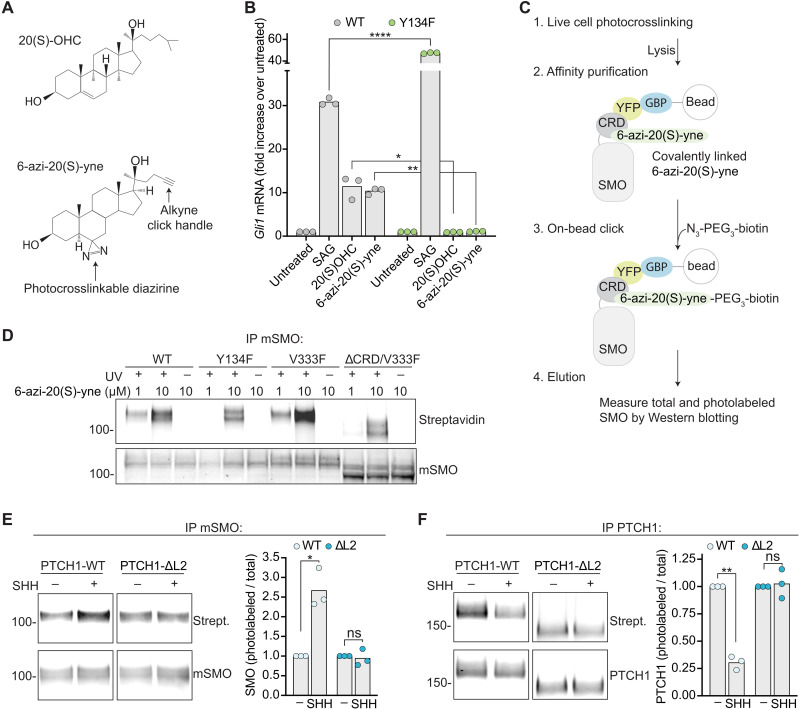
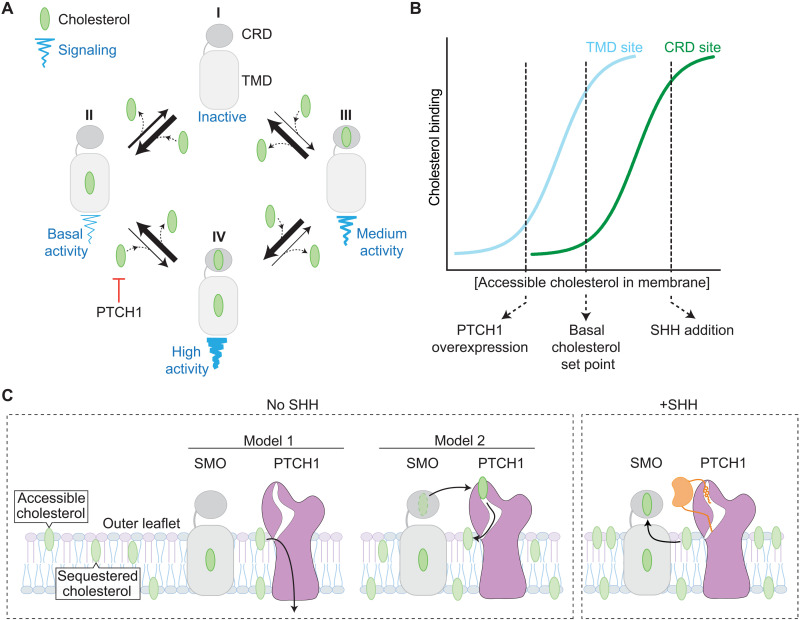
Smoothened (SMO) transduces the Hedgehog (Hh) signal across the plasma membrane in response to accessible cholesterol. Cholesterol binds SMO at two sites: one in the extracellular cysteine-rich domain (CRD) and a second in the transmembrane domain (TMD). How these two sterol-binding sites mediate SMO activation in response to the ligand Sonic Hedgehog (SHH) remains unknown. We find that mutations in the CRD (but not the TMD) reduce the fold increase in SMO activity triggered by SHH. SHH also promotes the photocrosslinking of a sterol analog to the CRD in intact cells. In contrast, sterol binding to the TMD site boosts SMO activity regardless of SHH exposure. Mutational and computational analyses show that these sites are in allosteric communication despite being 45 angstroms apart. Hence, sterols function as both SHH-regulated orthosteric ligands at the CRD and allosteric ligands at the TMD to regulate SMO activity and Hh signaling.
Figures
References
-
- Kinnebrew M., Iverson E. J., Patel B. B., Pusapati G. V., Kong J. H., Johnson K. A., Luchetti G., Eckert K. M., McDonald J. G., Covey D. F., Siebold C., Radhakrishnan A., Rohatgi R., Cholesterol accessibility at the ciliary membrane controls hedgehog signaling. eLife 8, e50051 (2019). - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
