

BCL10 Mutations Define Distinct Dependencies Guiding Precision Therapy for DLBCL
- PMID: 35658124
- PMCID: PMC9357155
- DOI: 10.1158/2159-8290.CD-21-1566
BCL10 Mutations Define Distinct Dependencies Guiding Precision Therapy for DLBCL
Abstract
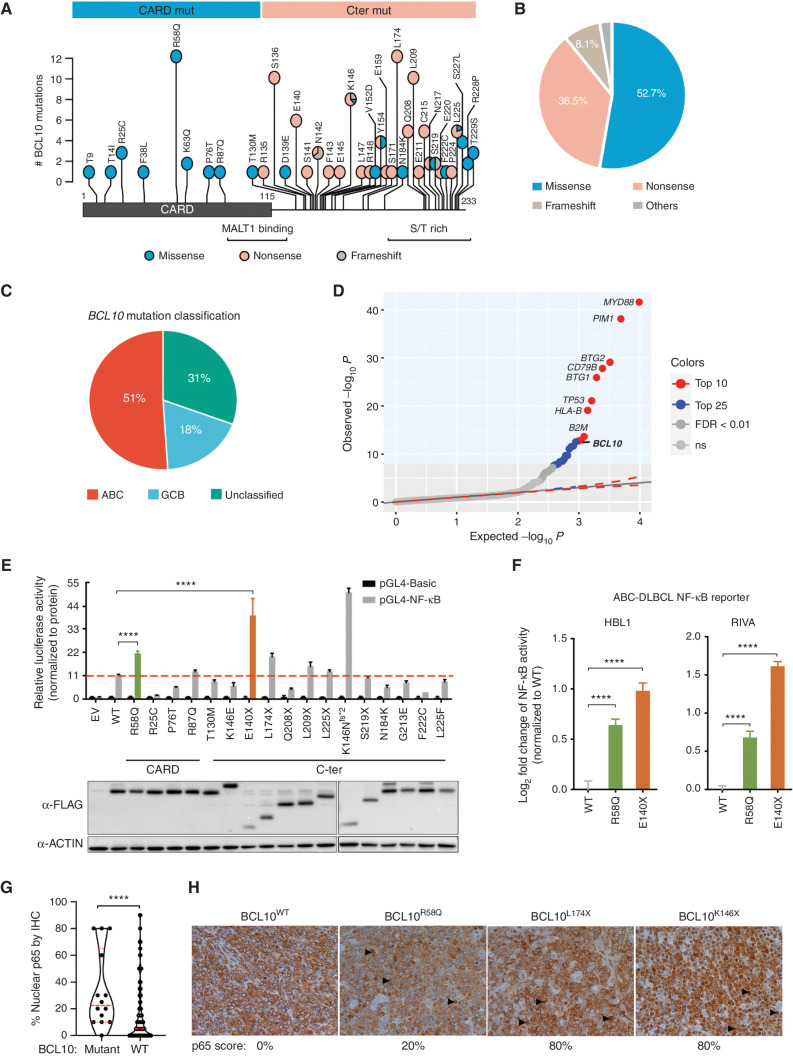
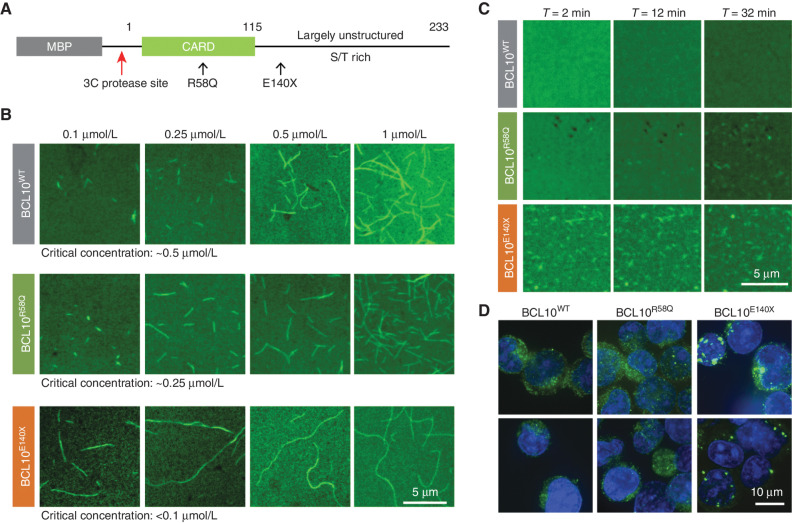
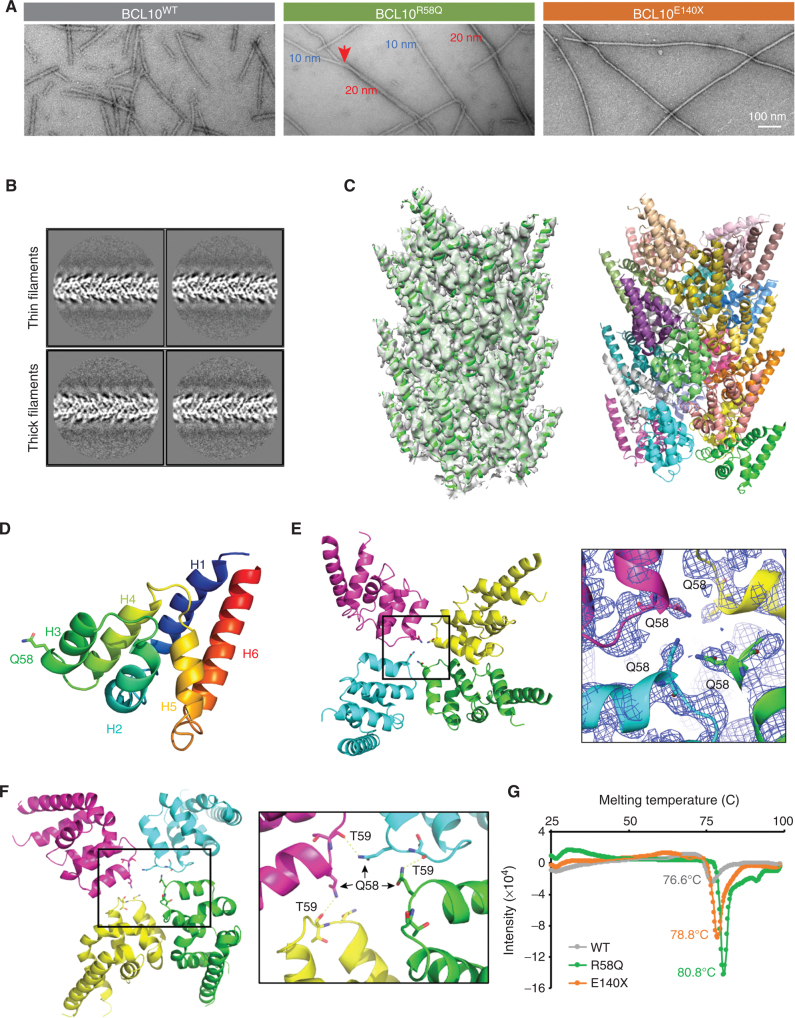
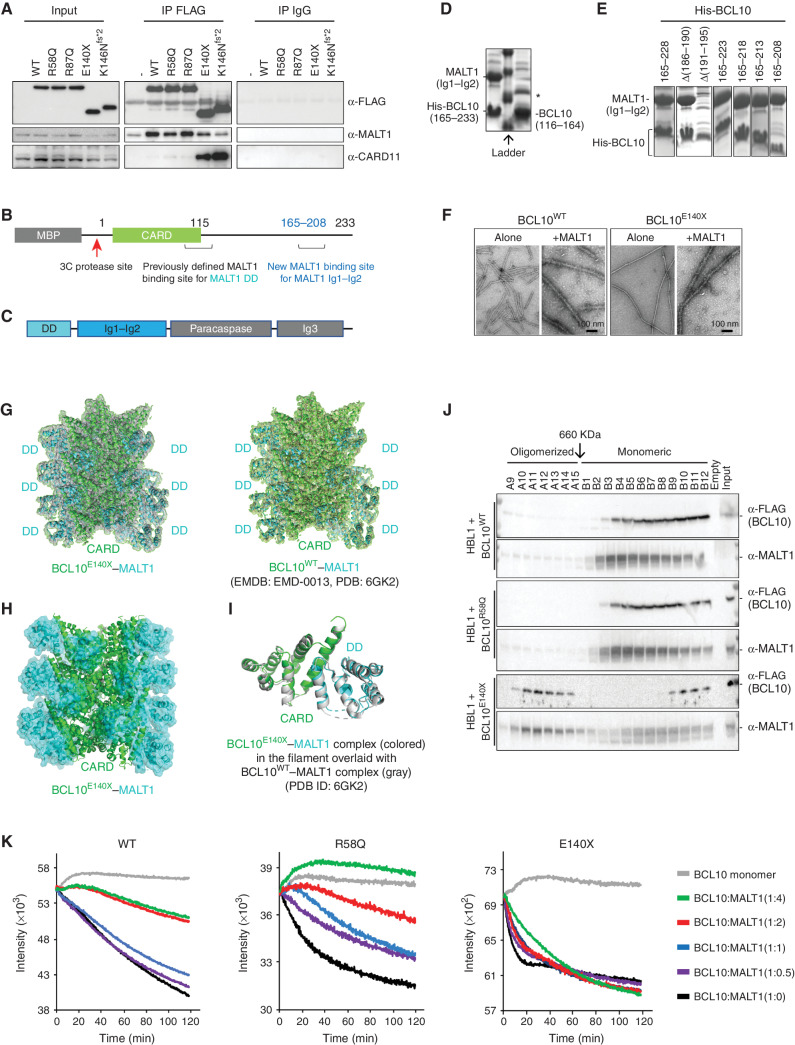
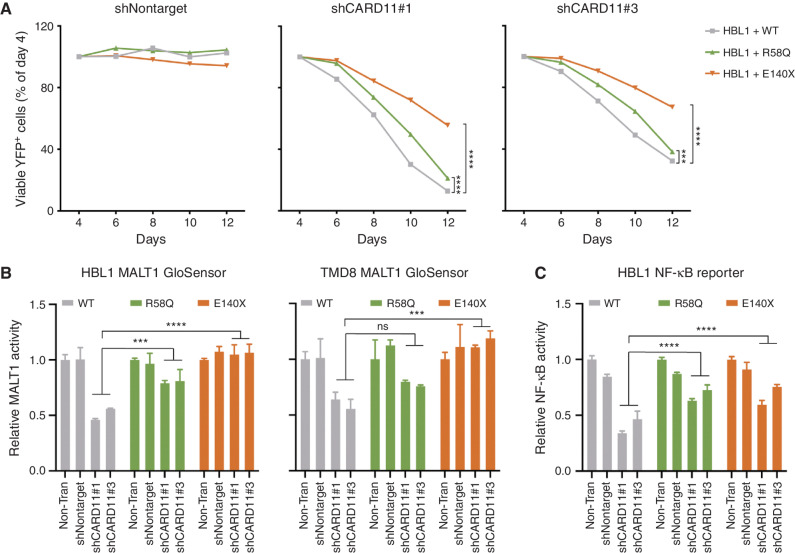
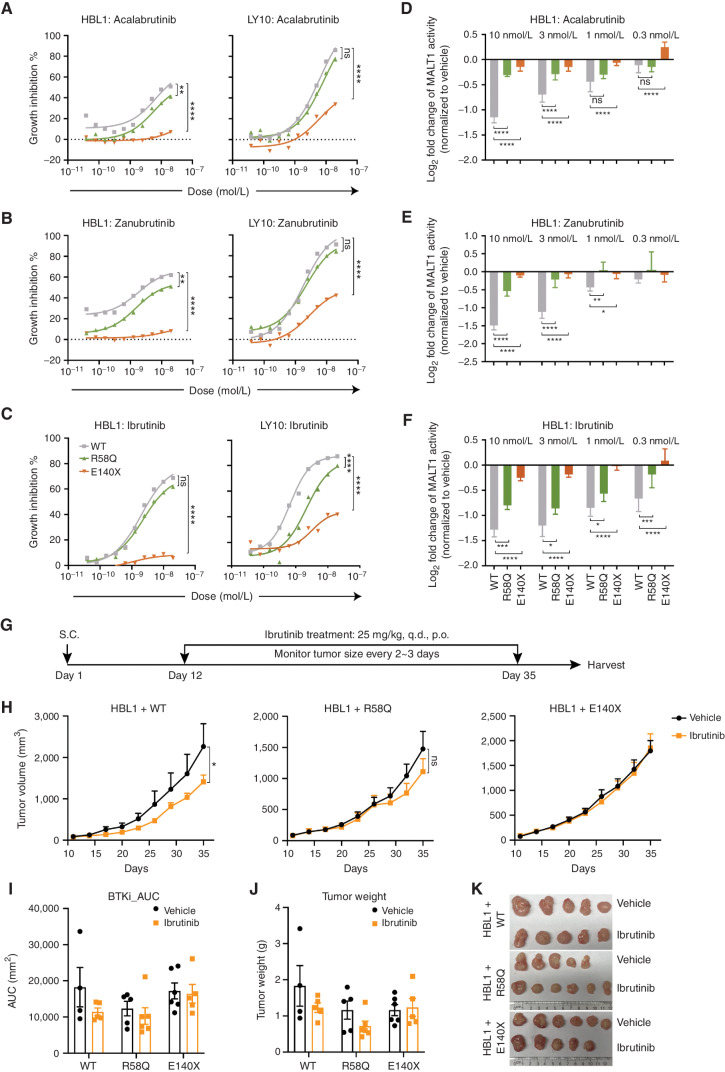
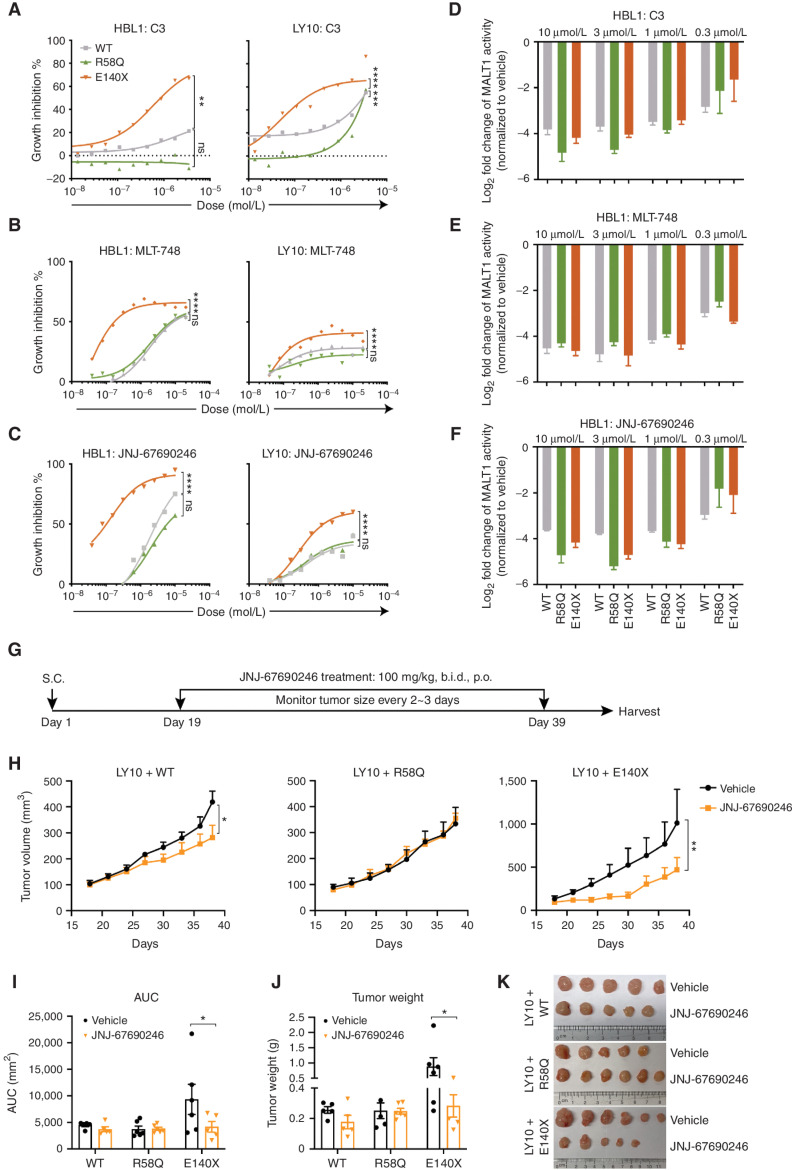
Activated B cell-like diffuse large B-cell lymphomas (ABC-DLBCL) have unfavorable outcomes and chronic activation of CARD11-BCL10-MALT1 (CBM) signal amplification complexes that form due to polymerization of BCL10 subunits, which is affected by recurrent somatic mutations in ABC-DLBCLs. Herein, we show that BCL10 mutants fall into at least two functionally distinct classes: missense mutations of the BCL10 CARD domain and truncation of its C-terminal tail. Truncating mutations abrogated a motif through which MALT1 inhibits BCL10 polymerization, trapping MALT1 in its activated filament-bound state. CARD missense mutations enhanced BCL10 filament formation, forming glutamine network structures that stabilize BCL10 filaments. Mutant forms of BCL10 were less dependent on upstream CARD11 activation and thus manifested resistance to BTK inhibitors, whereas BCL10 truncating but not CARD mutants were hypersensitive to MALT1 inhibitors. Therefore, BCL10 mutations are potential biomarkers for BTK inhibitor resistance in ABC-DLBCL, and further precision can be achieved by selecting therapy based on specific biochemical effects of distinct mutation classes.
Significance: ABC-DLBCLs feature frequent mutations of signaling mediators that converge on the CBM complex. We use structure-function approaches to reveal that BCL10 mutations fall into two distinct biochemical classes. Both classes confer resistance to BTK inhibitors, whereas BCL10 truncations confer hyperresponsiveness to MALT1 inhibitors, providing a road map for precision therapies in ABC-DLBCLs. See related commentary by Phelan and Oellerich, p. 1844. This article is highlighted in the In This Issue feature, p. 1825.
©2022 The Authors; Published by the American Association for Cancer Research.
Figures
Comment in
-
BCL10 Mutants: Architects of Oncogenic Signaling Provide a Blueprint for Precision Medicine.Cancer Discov. 2022 Aug 5;12(8):1844-1846. doi: 10.1158/2159-8290.CD-22-0614. Cancer Discov. 2022. PMID: 35929131
References
-
- Meininger I, Krappmann D. Lymphocyte signaling and activation by the CARMA1-BCL10-MALT1 signalosome. Biol Chem 2016;397:1315–33. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
