

Insertion-and-Deletion Mutations between the Genomes of SARS-CoV, SARS-CoV-2, and Bat Coronavirus RaTG13
- PMID: 35658573
- PMCID: PMC9241832
- DOI: 10.1128/spectrum.00716-22
Insertion-and-Deletion Mutations between the Genomes of SARS-CoV, SARS-CoV-2, and Bat Coronavirus RaTG13
Abstract
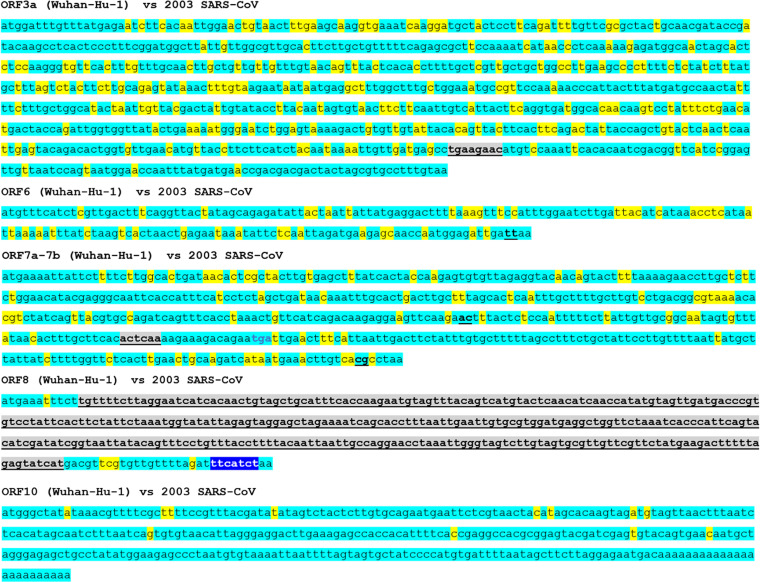
The evolutional process of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) development remains inconclusive. This study compared the genome sequences of severe acute respiratory syndrome coronavirus (SARS-CoV), bat coronavirus RaTG13, and SARS-CoV-2. In total, the genomes of SARS-CoV-2 and RaTG13 were 77.9% and 77.7% identical to the genome of SARS-CoV, respectively. A total of 3.6% (1,068 bases) of the SARS-CoV-2 genome was derived from insertion and/or deletion (indel) mutations, and 18.6% (5,548 bases) was from point mutations from the genome of SARS-CoV. At least 35 indel sites were confirmed in the genome of SARS-CoV-2, in which 17 were with ≥10 consecutive bases long. Ten of these relatively long indels were located in the spike (S) gene, five in nonstructural protein 3 (Nsp3) gene of open reading frame (ORF) 1a, and one in ORF8 and noncoding region. Seventeen (48.6%) of the 35 indels were based on insertion-and-deletion mutations with exchanged gene sequences of 7-325 consecutive bases. Almost the complete ORF8 gene was replaced by a single 325 consecutive base-long indel. The distribution of these indels was roughly in accordance with the distribution of the rate of point mutation rate around the indels. The genome sequence of SARS-CoV-2 was 96.0% identical to that of RaTG13. There was no long insertion-and-deletion mutation between the genomes of RaTG13 and SARS-CoV-2. The findings of the uneven distribution of multiple indels and the presence of multiple long insertion-and-deletion mutations with exchanged consecutive base sequences in the viral genome may provide insights into SARS-CoV-2 development. IMPORTANCE The developmental mechanism of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) remains inconclusive. This study compared the base sequence one-by-one between severe acute respiratory syndrome coronavirus (SARS-CoV) or bat coronavirus RaTG13 and SARS-CoV-2. The genomes of SARS-CoV-2 and RaTG13 were 77.9% and 77.7% identical to the genome of SARS-CoV, respectively. Seventeen of the 35 sites with insertion and/or deletion mutations between SARS-CoV-2 and SARS-CoV were based on insertion-and-deletion mutations with the replacement of 7-325 consecutive bases. Most of these long insertion-and-deletion sites were concentrated in the nonstructural protein 3 (Nsp3) gene of open reading frame (ORF) 1a, S1 domain of the spike protein, and ORF8 genes. Such long insertion-and-deletion mutations were not observed between the genomes of RaTG13 and SARS-CoV-2. The presence of multiple long insertion-and-deletion mutations in the genome of SARS-CoV-2 and their uneven distributions may provide further insights into the development of the virus.
Keywords: bat coronavirus RaTG13; coronavirus disease 2019 (COVID-19); insertion-and-deletion mutation; mutation; severe acute respiratory syndrome coronavirus (SARS-CoV); severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2).
Conflict of interest statement
The authors declare no conflict of interest.
Figures







References
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
