

Apolipoprotein A-I, elevated in trauma patients, inhibits platelet activation and decreases clot strength
- PMID: 35659185
- PMCID: PMC9547822
- DOI: 10.1080/09537104.2022.2078488
Apolipoprotein A-I, elevated in trauma patients, inhibits platelet activation and decreases clot strength
Abstract
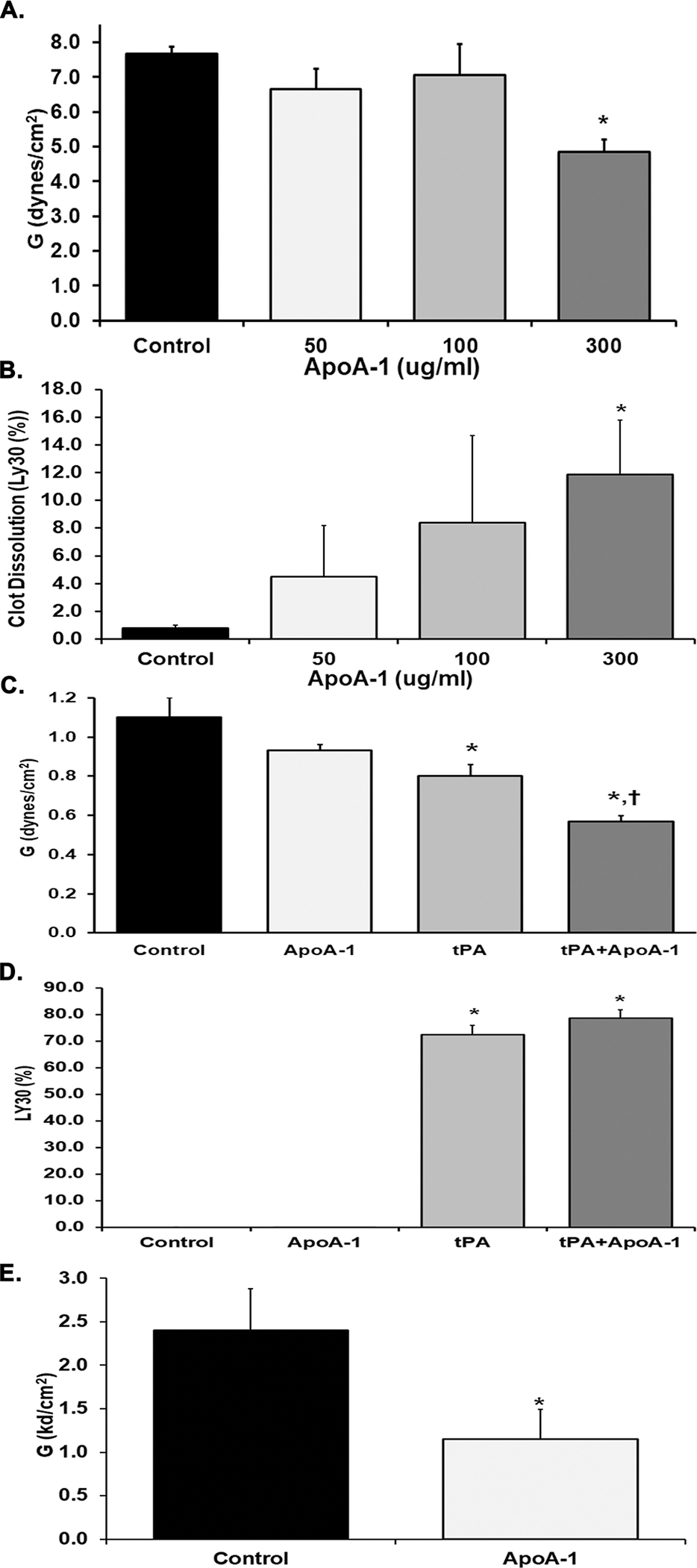
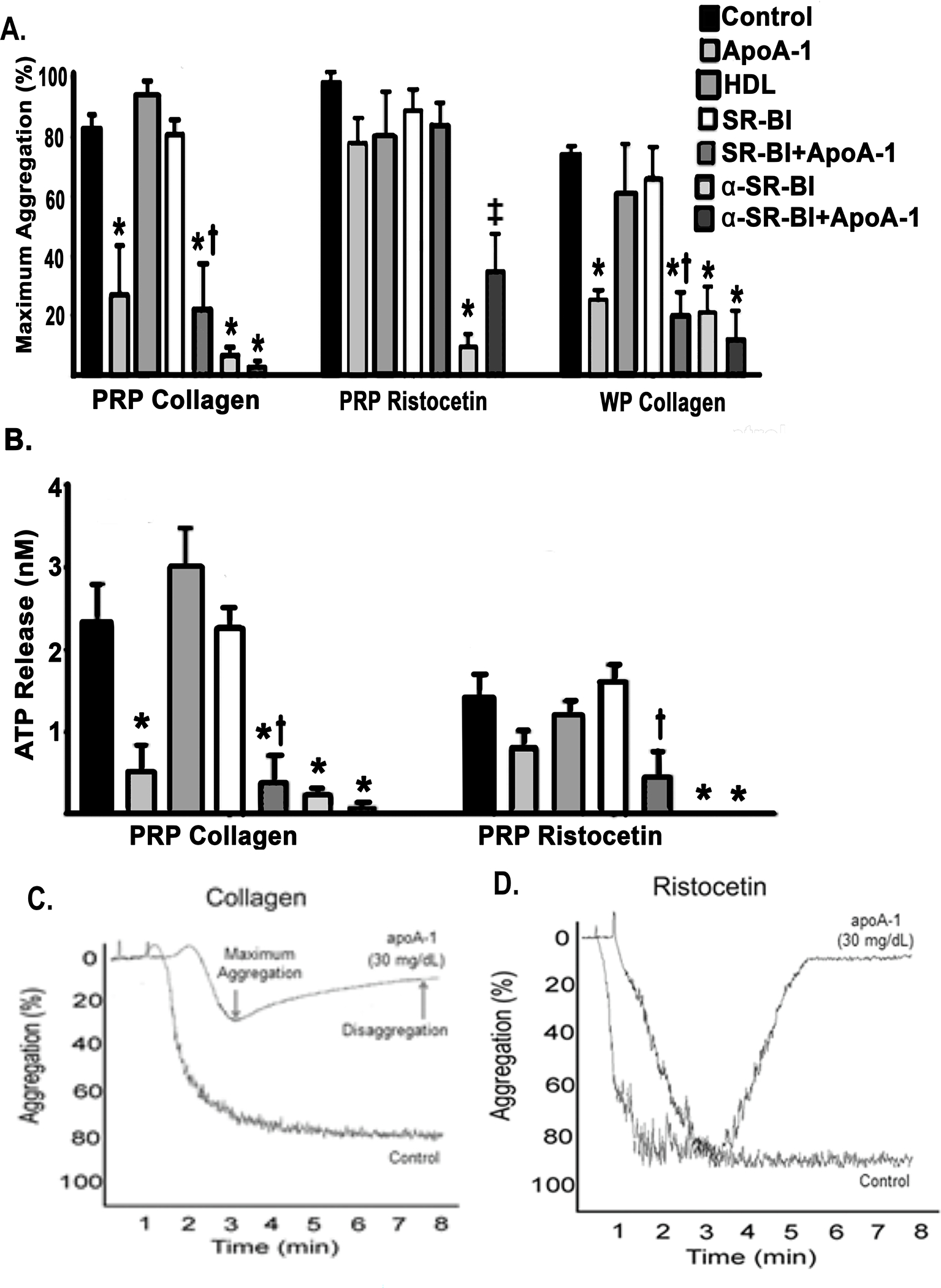
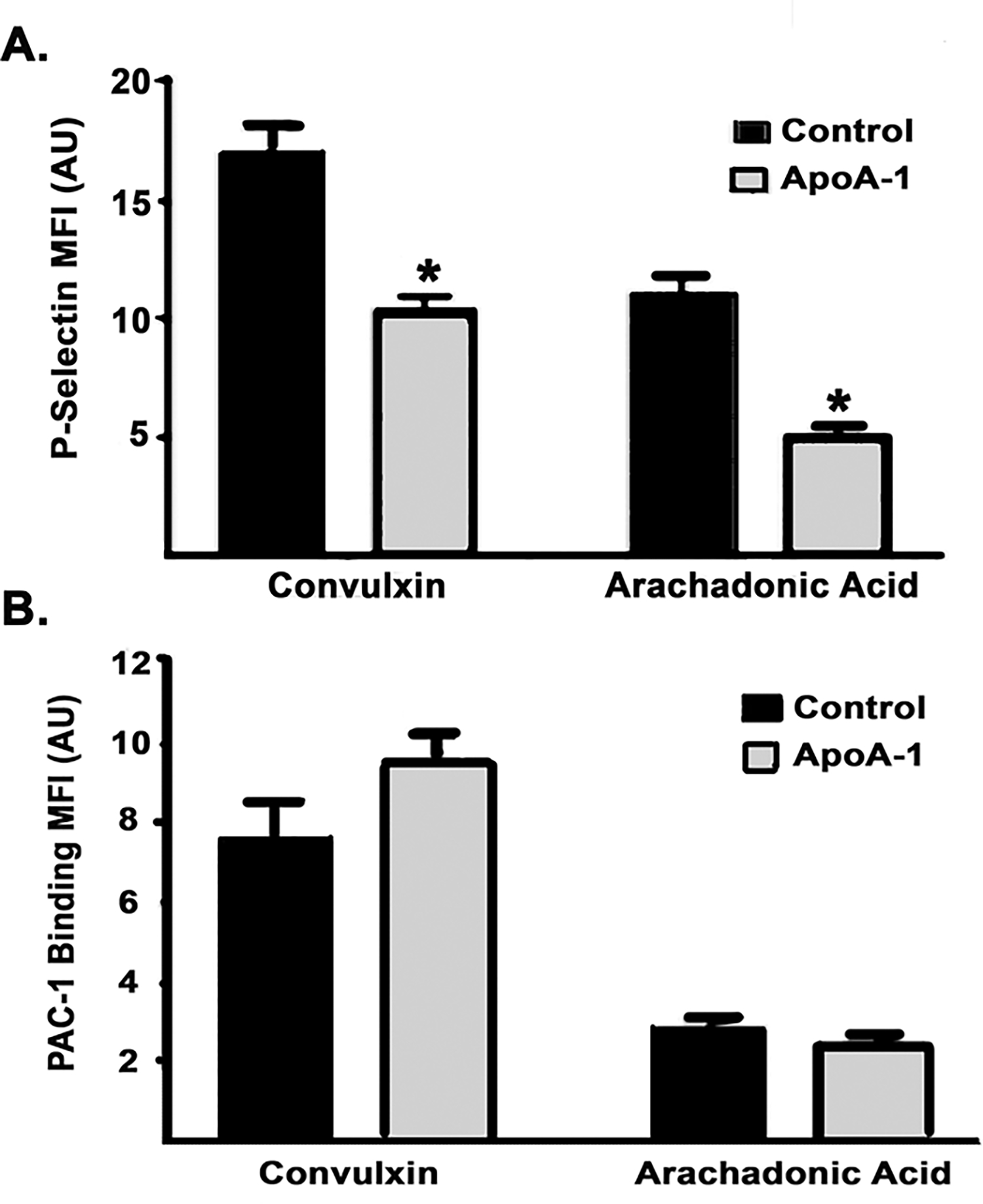
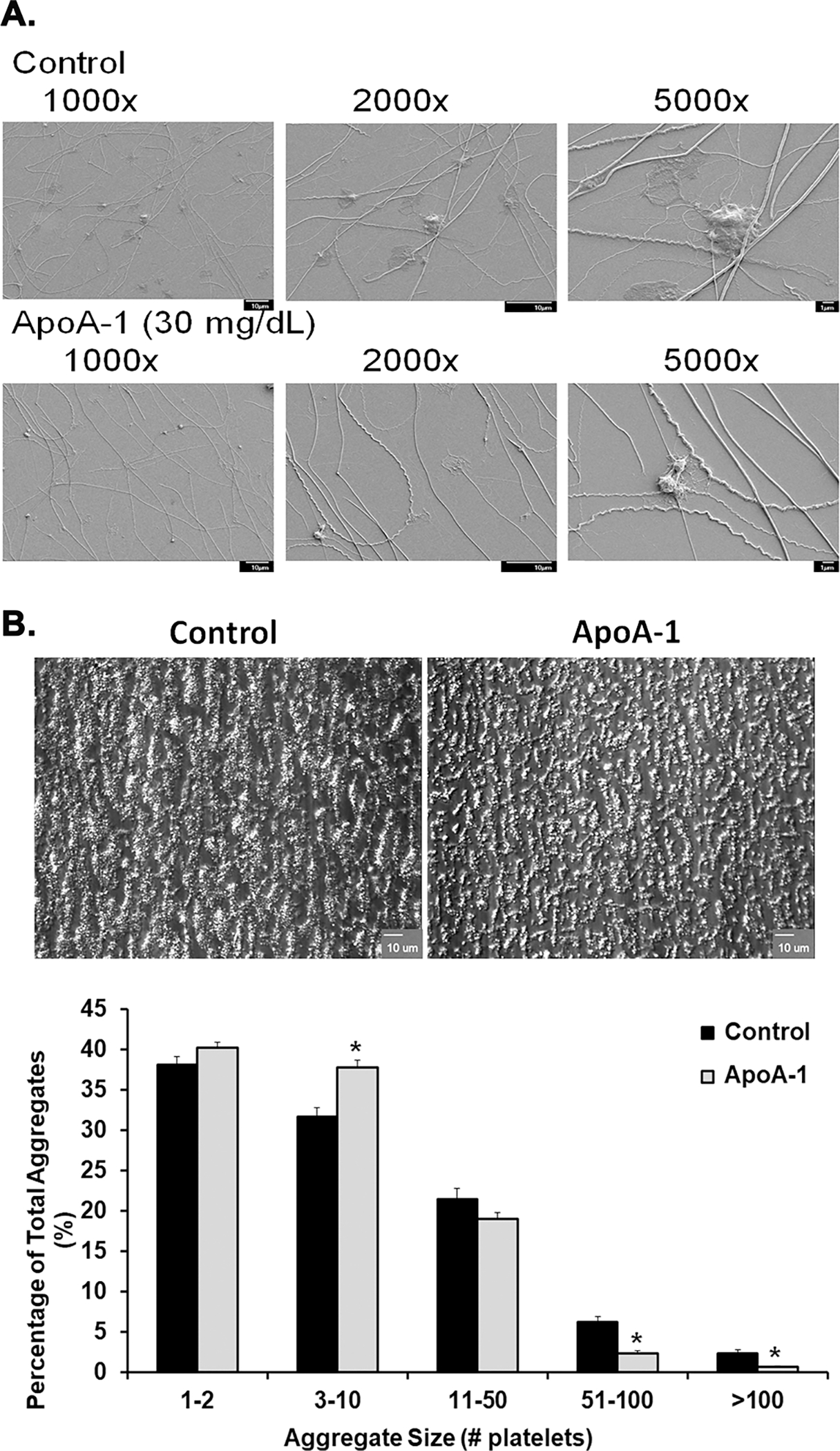
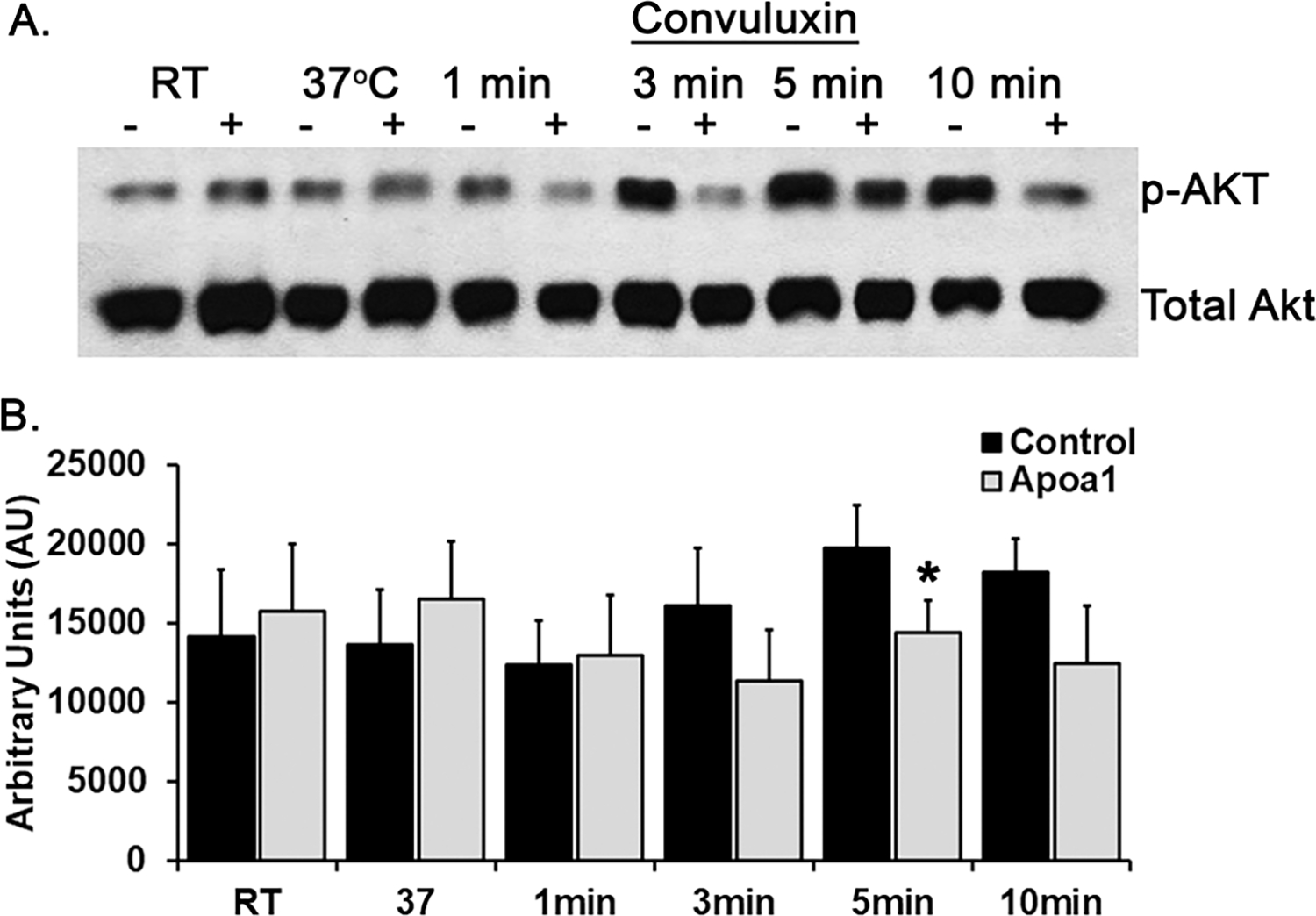
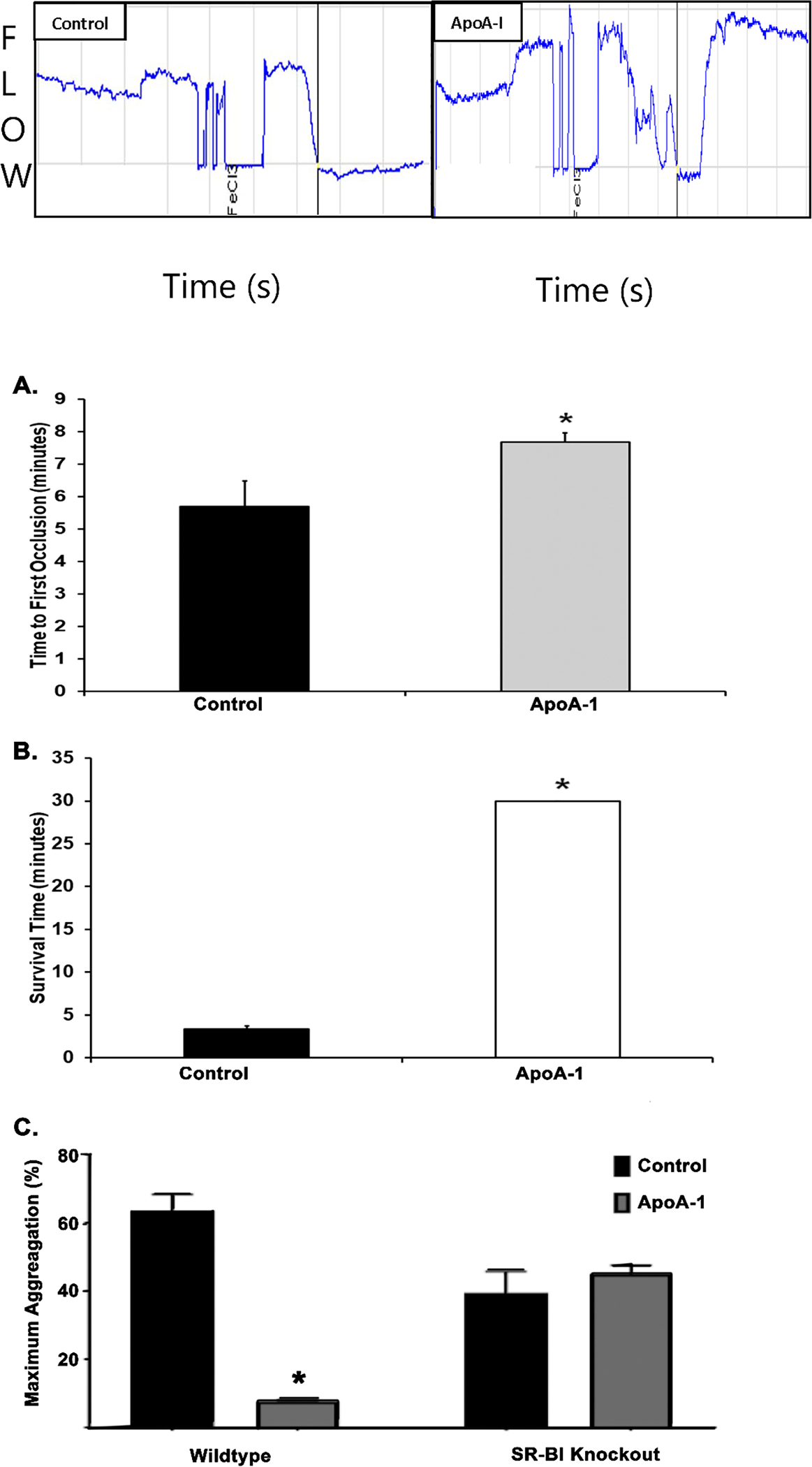
Apolipoprotein A-I (ApoA-I) is elevated in the plasma of a subgroup of trauma patients with systemic hyperfibrinolysis. We hypothesize that apoA-I inhibits platelet activation and clot formation. The effects of apoA-I on human platelet activation and clot formation were assessed by whole blood thrombelastography (TEG), platelet aggregometry, P-selectin surface expression, microfluidic adhesion, and Akt phosphorylation. Mouse models of carotid artery thrombosis and pulmonary embolism were used to assess the effects of apoA-I in vivo. The ApoA-1 receptor was investigated with transgenic mice knockouts (KO) for the scavenger receptor class B member 1 (SR-BI). Compared to controls, exogenous human apoA-I inhibited arachidonic acid and collagen-mediated human and mouse platelet aggregation, decreased P-selectin surface expression and Akt activation, resulting in diminished clot strength and increased clot lysis by TEG. ApoA-I also decreased platelet aggregate size formed on a collagen surface under flow. In vivo, apoA-I delayed vessel occlusion in an arterial thrombosis model and conferred a survival advantage in a pulmonary embolism model. SR-BI KO mice significantly reduced apoA-I inhibition of platelet aggregation versus wild-type platelets. Exogenous human apoA-I inhibits platelet activation, decreases clot strength and stability, and protects mice from arterial and venous thrombosis via the SR-BI receptor.
Keywords: Hyperfibrinolysis; SR-B1 receptor; microfluidics; platelet inhibition; thrombelastography.
Conflict of interest statement
Disclosure Statement
No authors have relevant conflicts of interest to declare.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous