

Humanized yeast to model human biology, disease and evolution
- PMID: 35661208
- PMCID: PMC9194483
- DOI: 10.1242/dmm.049309
Humanized yeast to model human biology, disease and evolution
Abstract
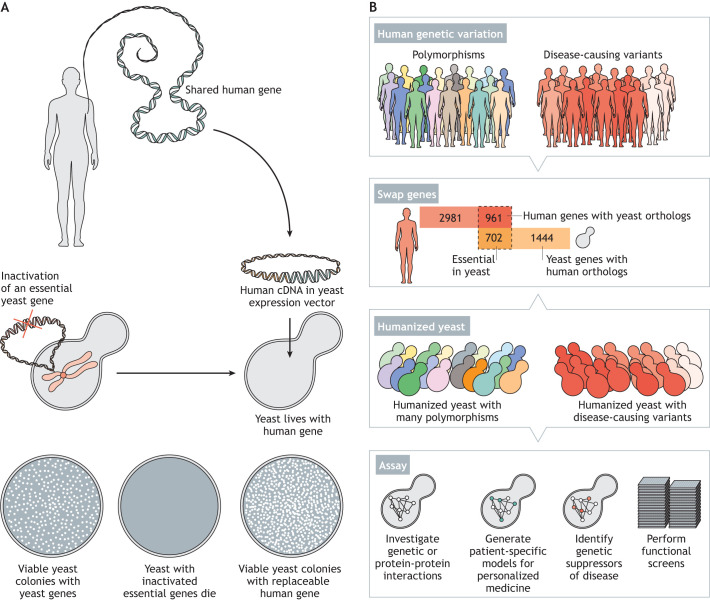
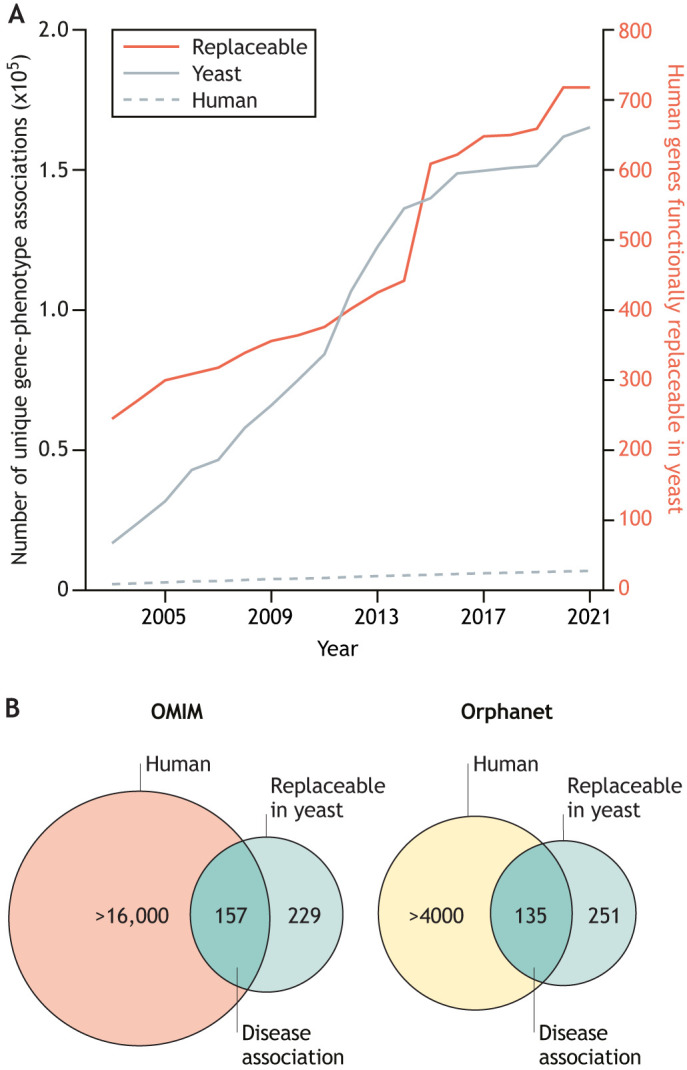
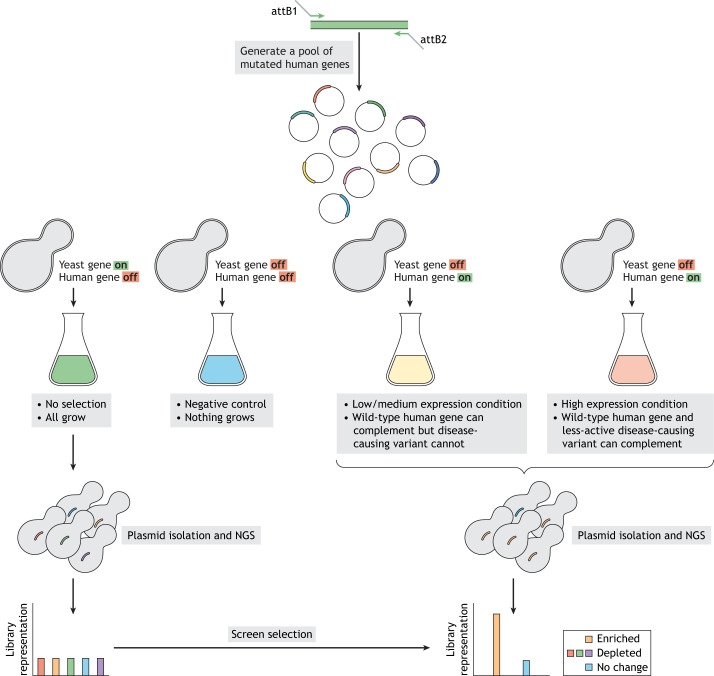
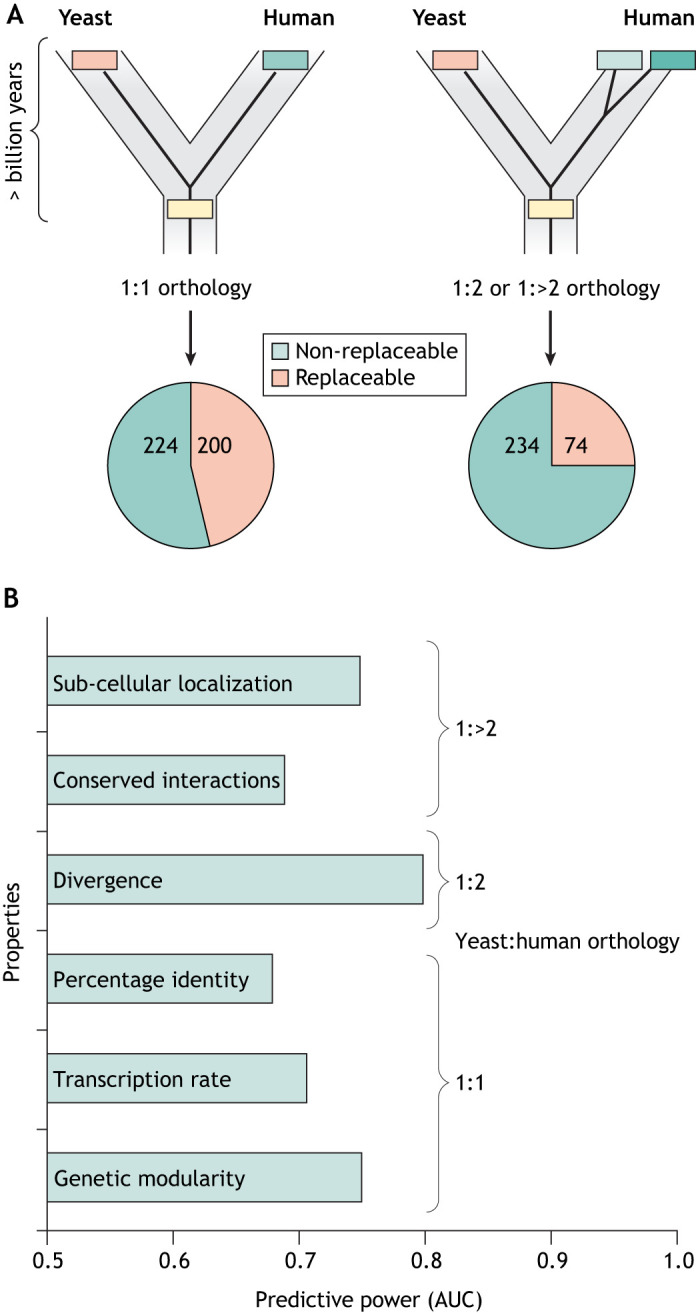
For decades, budding yeast, a single-cellular eukaryote, has provided remarkable insights into human biology. Yeast and humans share several thousand genes despite morphological and cellular differences and over a billion years of separate evolution. These genes encode critical cellular processes, the failure of which in humans results in disease. Although recent developments in genome engineering of mammalian cells permit genetic assays in human cell lines, there is still a need to develop biological reagents to study human disease variants in a high-throughput manner. Many protein-coding human genes can successfully substitute for their yeast equivalents and sustain yeast growth, thus opening up doors for developing direct assays of human gene function in a tractable system referred to as 'humanized yeast'. Humanized yeast permits the discovery of new human biology by measuring human protein activity in a simplified organismal context. This Review summarizes recent developments showing how humanized yeast can directly assay human gene function and explore variant effects at scale. Thus, by extending the 'awesome power of yeast genetics' to study human biology, humanizing yeast reinforces the high relevance of evolutionarily distant model organisms to explore human gene evolution, function and disease.
Keywords: Functional complementation; Functional replaceability; Humanized yeast; Orthology.
© 2022. Published by The Company of Biologists Ltd.
Conflict of interest statement
Competing interests The authors declare no competing or financial interests.
Figures

References
-
- 1000 Genomes Project Consortium, Auton, A., Brooks, L. D., Durbin, R. M., Garrison, E. P., Kang, H. M., Korbel, J. O., Marchini, J. L., McCarthy, S., McVean, G. A., and Abecasis, G. R. (2015). A global reference for human genetic variation. Nature 526, 68-74. 10.1038/nature15393 - DOI - PMC - PubMed
-
- Ahn, J., Choi, C.-H., Kang, C.-M., Kim, C.-H., Park, H.-M., Song, K.-B., Hoe, K.-L., Won, M. and Chung, K.-S. (2009). Generation of expression vectors for high-throughput functional analysis of target genes in Schizosaccharomyces pombe. J. Microbiol. 47, 789-795. 10.1007/s12275-009-0010-4 - DOI - PubMed
-
- Aiyar, R. S., Bohnert, M., Duvezin-Caubet, S., Voisset, C., Gagneur, J., Fritsch, E. S., Couplan, E., von der Malsburg, K., Funaya, C., Soubigou, F.et al. (2014). Mitochondrial protein sorting as a therapeutic target for ATP synthase disorders. Nat. Commun. 5, 5585. 10.1038/ncomms6585 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
