

Identification of Synergistic Drug Combinations to Target KRAS-Driven Chemoradioresistant Cancers Utilizing Tumoroid Models of Colorectal Adenocarcinoma and Recurrent Glioblastoma
- PMID: 35664781
- PMCID: PMC9158132
- DOI: 10.3389/fonc.2022.840241
Identification of Synergistic Drug Combinations to Target KRAS-Driven Chemoradioresistant Cancers Utilizing Tumoroid Models of Colorectal Adenocarcinoma and Recurrent Glioblastoma
Abstract
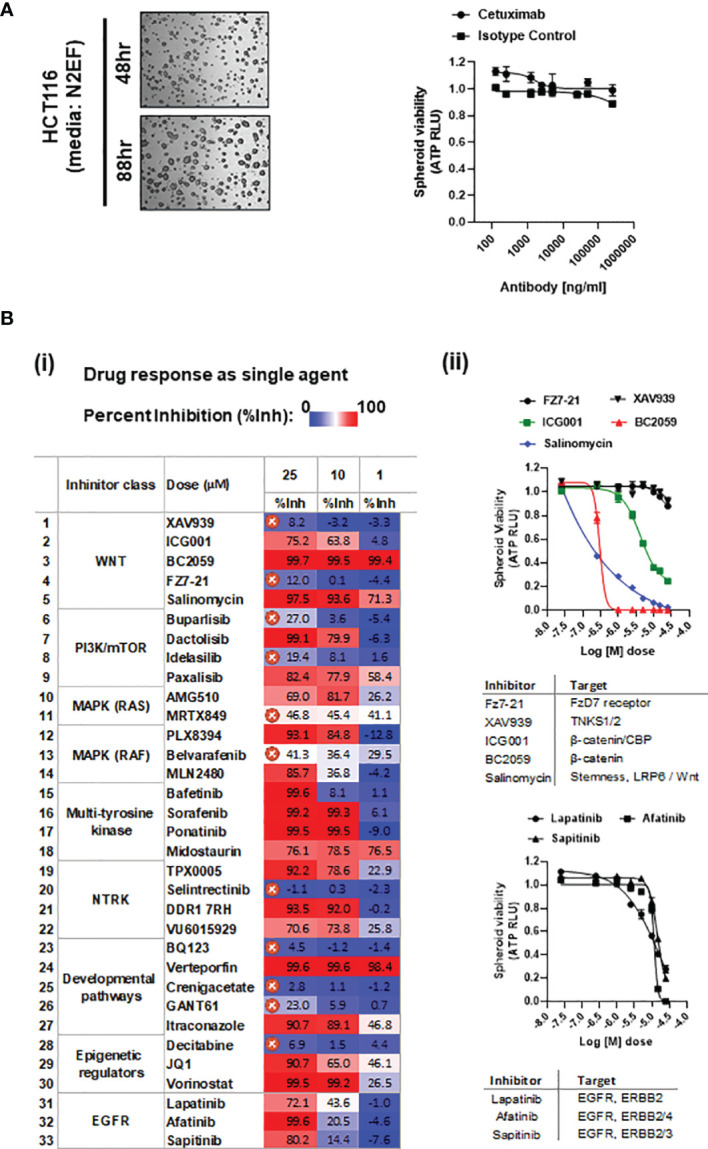
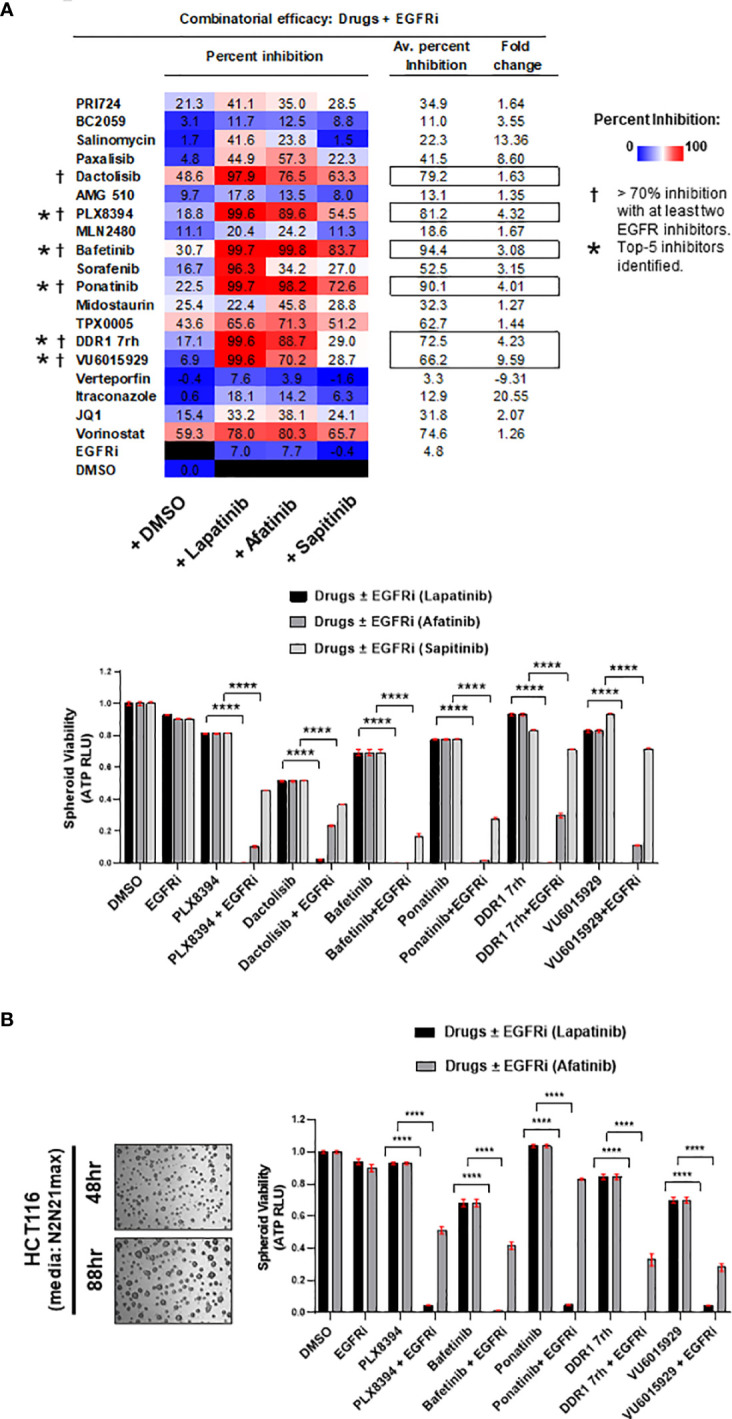
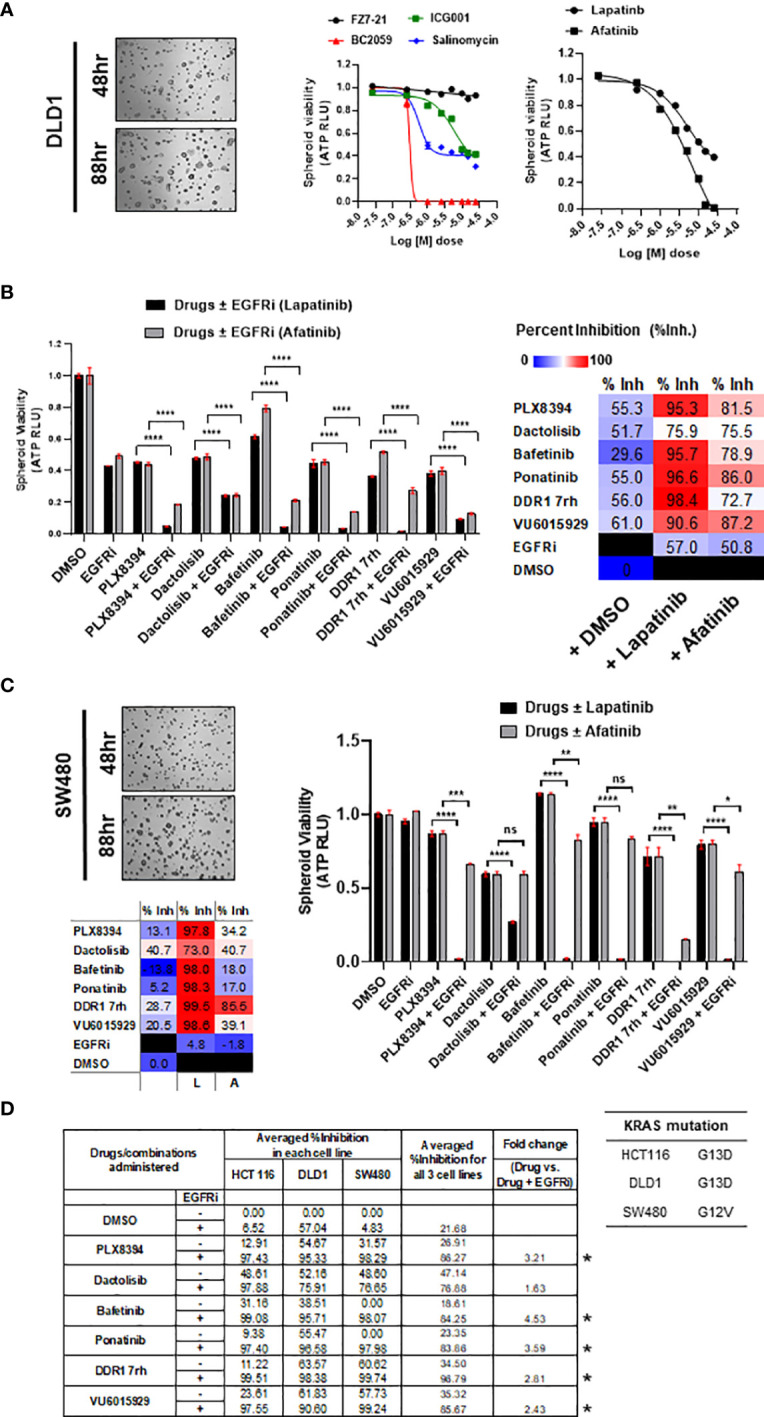
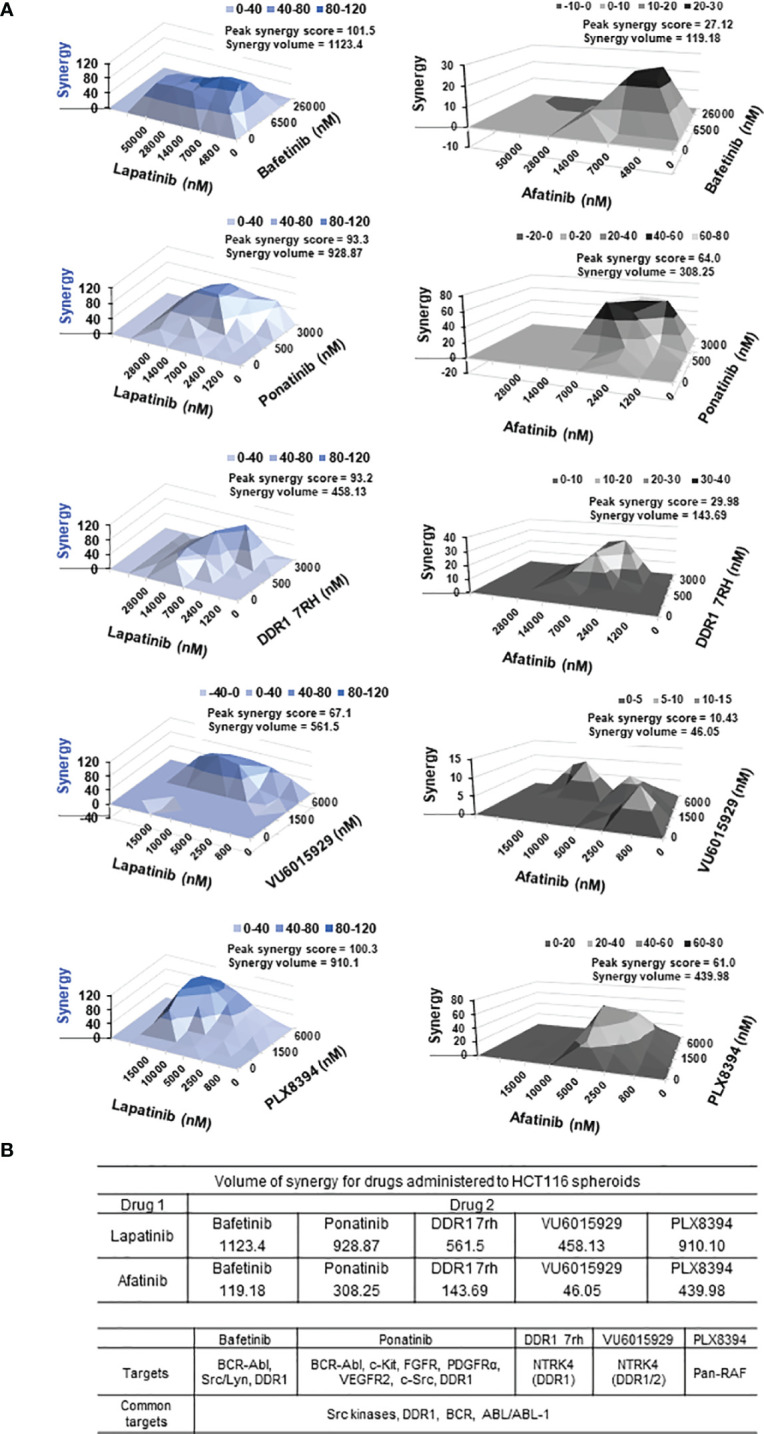
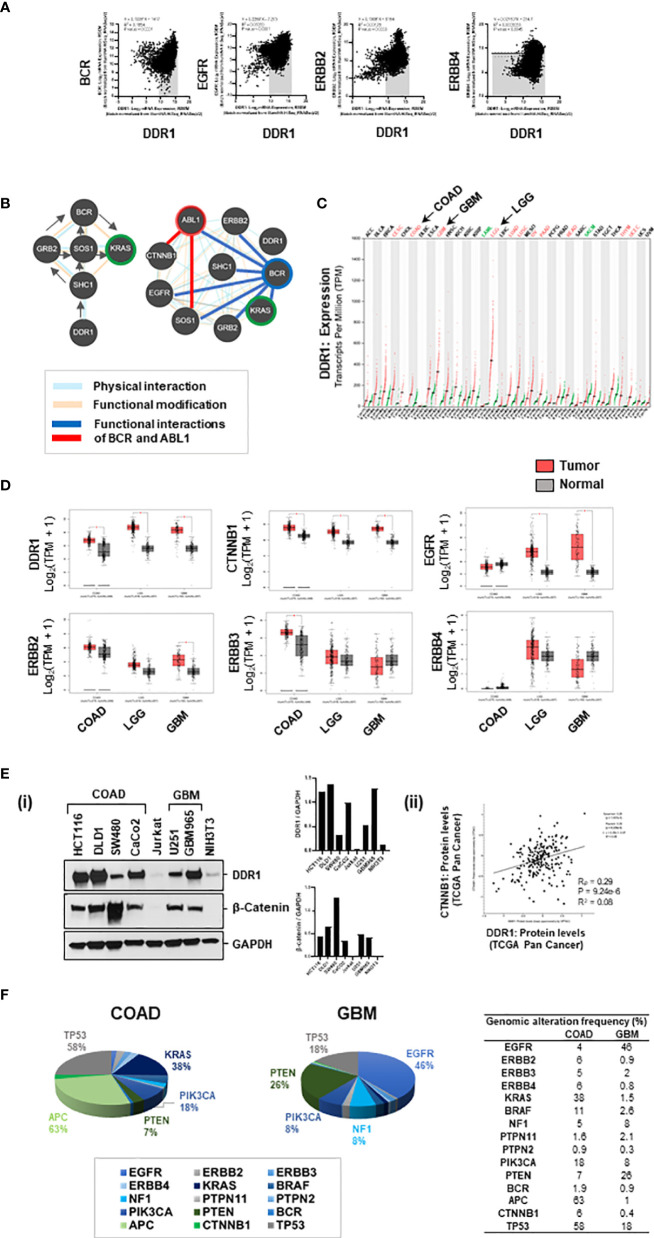
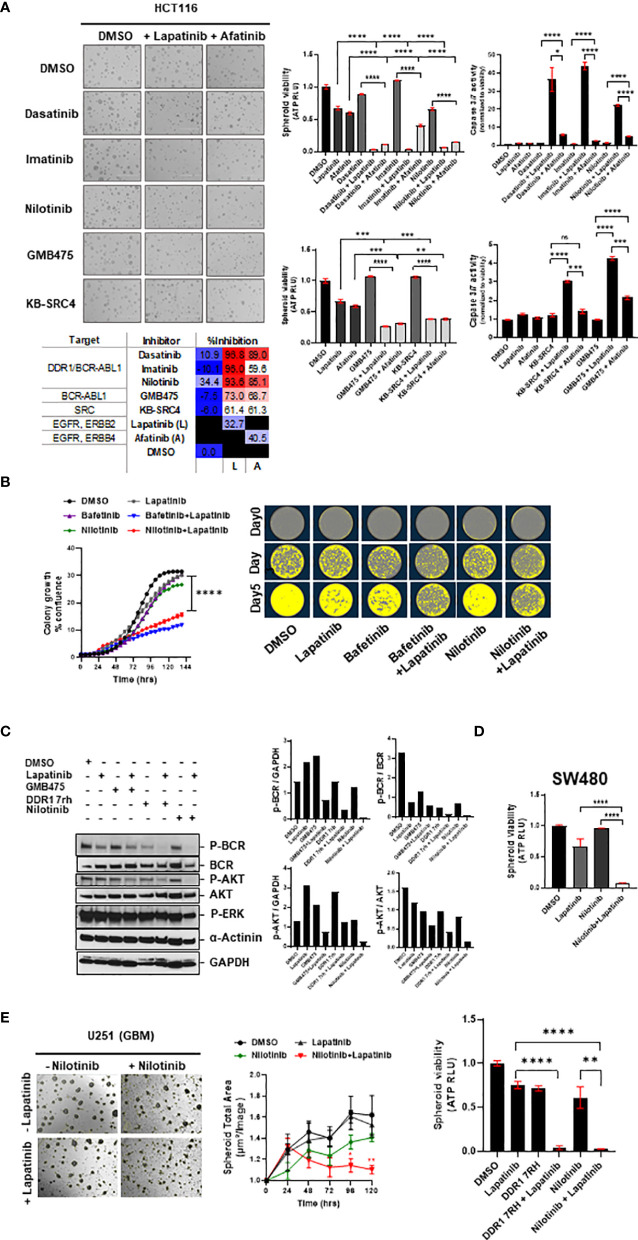
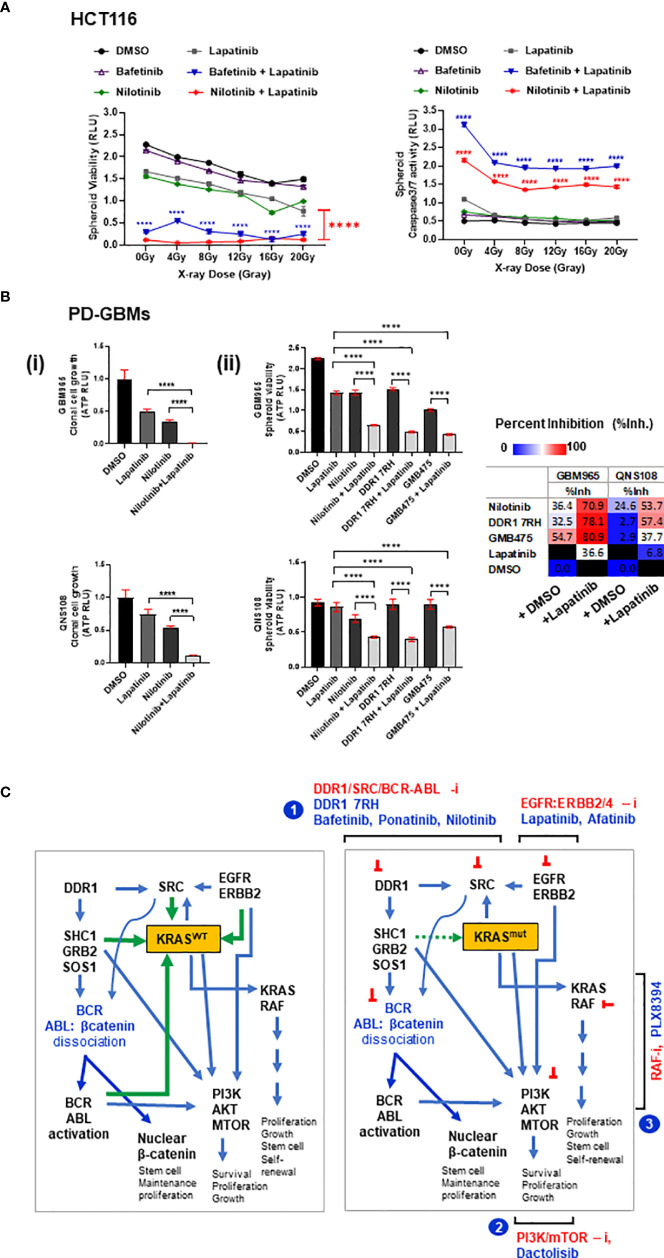
Treatment resistance is observed in all advanced cancers. Colorectal cancer (CRC) presenting as colorectal adenocarcinoma (COAD) is the second leading cause of cancer deaths worldwide. Multimodality treatment includes surgery, chemotherapy, and targeted therapies with selective utilization of immunotherapy and radiation therapy. Despite the early success of anti-epidermal growth factor receptor (anti-EGFR) therapy, treatment resistance is common and often driven by mutations in APC, KRAS, RAF, and PI3K/mTOR and positive feedback between activated KRAS and WNT effectors. Challenges in the direct targeting of WNT regulators and KRAS have caused alternative actionable targets to gain recent attention. Utilizing an unbiased drug screen, we identified combinatorial targeting of DDR1/BCR-ABL signaling axis with small-molecule inhibitors of EGFR-ERBB2 to be potentially cytotoxic against multicellular spheroids obtained from WNT-activated and KRAS-mutant COAD lines (HCT116, DLD1, and SW480) independent of their KRAS mutation type. Based on the data-driven approach using available patient datasets (The Cancer Genome Atlas (TCGA)), we constructed transcriptomic correlations between gene DDR1, with an expression of genes for EGFR, ERBB2-4, mitogen-activated protein kinase (MAPK) pathway intermediates, BCR, and ABL and genes for cancer stem cell reactivation, cell polarity, and adhesion; we identified a positive association of DDR1 with EGFR, ERBB2, BRAF, SOX9, and VANGL2 in Pan-Cancer. The evaluation of the pathway network using the STRING database and Pathway Commons database revealed DDR1 protein to relay its signaling via adaptor proteins (SHC1, GRB2, and SOS1) and BCR axis to contribute to the KRAS-PI3K-AKT signaling cascade, which was confirmed by Western blotting. We further confirmed the cytotoxic potential of our lead combination involving EGFR/ERBB2 inhibitor (lapatinib) with DDR1/BCR-ABL inhibitor (nilotinib) in radioresistant spheroids of HCT116 (COAD) and, in an additional devastating primary cancer model, glioblastoma (GBM). GBMs overexpress DDR1 and share some common genomic features with COAD like EGFR amplification and WNT activation. Moreover, genetic alterations in genes like NF1 make GBMs have an intrinsically high KRAS activity. We show the combination of nilotinib plus lapatinib to exhibit more potent cytotoxic efficacy than either of the drugs administered alone in tumoroids of patient-derived recurrent GBMs. Collectively, our findings suggest that combinatorial targeting of DDR1/BCR-ABL with EGFR-ERBB2 signaling may offer a therapeutic strategy against stem-like KRAS-driven chemoradioresistant tumors of COAD and GBM, widening the window for its applications in mainstream cancer therapeutics.
Keywords: BCR-ABL1; COAD; DDR1; EGFR-ERBB2; GBM; KRAS; Wnt/b-catenin; chemoradioresistance.
Copyright © 2022 Gupta, Jones, Farias, Mackeyev, Singh, Quiñones-Hinojosa and Krishnan.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous