

Rothmund-Thomson syndrome type 1 caused by biallelic ANAPC1 gene mutations
- PMID: 35664819
- PMCID: PMC9060067
- DOI: 10.1002/ski2.12
Rothmund-Thomson syndrome type 1 caused by biallelic ANAPC1 gene mutations
Abstract
Background: Rare syndromic skin disorders may represent a diagnostic challenge.
Aims: We report a unique case associating cutaneous manifestations and developmental delay.
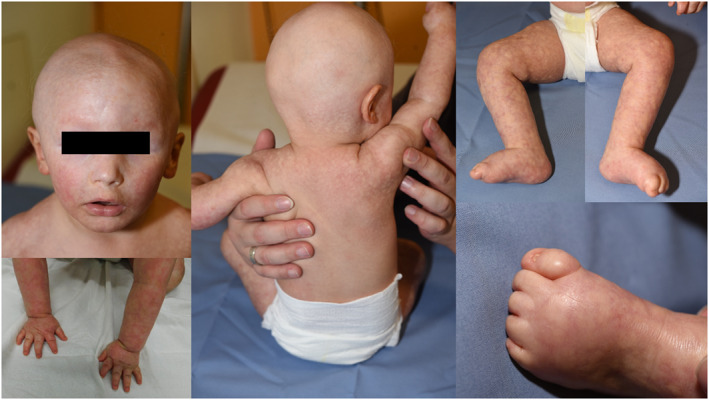
Materials & methods: The affected 14 months old boy had poikiloderma, facial dysmorphism with deep-set eyes, atrichia, as well as nail dysplasia and non-descended testes. In addition, his psychomotor development was delayed. Exome sequencing and molecular karyotyping via array-CGH (oligo-array, 180k Agilent, design 22060) were performed.
Results: Mutations in RECQL4 (found in patients with RTS2) were first excluded. In the ANAPC1 gene, a novel combination of a recurrent intronic mutation (c.2705-198C>T) and a deletion of the second ANAPC1 allele was detected, thus confirming the clinical diagnosis of RTS1. The deletion on chromosome 2q13 comprised further genes and spanned 1,7 megabases. Heterozygous deletions in this region are known as 2q13 microdeletion syndrome and are associated with developmental delay, autism and facial dysmorphism.
Discussion: The genetic findings most probably explain both, the RTS1 features and the developmental delay. Genetic diagnosis in RTS is indispensable to confirm the specific subtype and its associated risks: juvenile cataracts are features of RTS1 (ANAPC1 gene), whereas a high risk of osteosarcoma is part of RTS2 (RECQL4 gene). Thus, the patient described here is at high risk for the development of juvenile cataracts and requires regular ophthalmologic examination.
Conclusion: This case report underlines the necessity of thorough clinical diagnosis prior to genetic diagnosis of RTS1, since the recurrent intronic ANAPC1 mutation is otherwise missed.
© 2021 The Authors. Skin Health and Disease published by John Wiley & Sons Ltd on behalf of British Association of Dermatologists.
Conflict of interest statement
The authors declare that there is no conflict of interests.
Figures
References
-
- Kitao S, Shimamoto A, Goto M, et al. Mutations in RECQL4 cause a subset of cases of Rothmund‐Thomson syndrome. Nat Genet. 1999;22:82–84. - PubMed
-
- Lu L, Jin W, Wang LL. RECQ DNA helicates and osteosarcoma. Adv Exp Med Biol. 2020;1258:37–54. - PubMed
-
- Wang LL, Plon SE. Rothmund‐Thomson syndrome In: MargaretPA, HollyHA, RobertaAP, StephanieEW, LoraJHB, KarenS, AnneA, editors. GeneReviews® [Internet]. Seattle, WA: University of Washington; 1999. pp. 1993–2020.
Publication types
LinkOut - more resources
Full Text Sources