

Predictors of human-infective RNA virus discovery in the United States, China, and Africa, an ecological study
- PMID: 35666108
- PMCID: PMC9278958
- DOI: 10.7554/eLife.72123
Predictors of human-infective RNA virus discovery in the United States, China, and Africa, an ecological study
Abstract
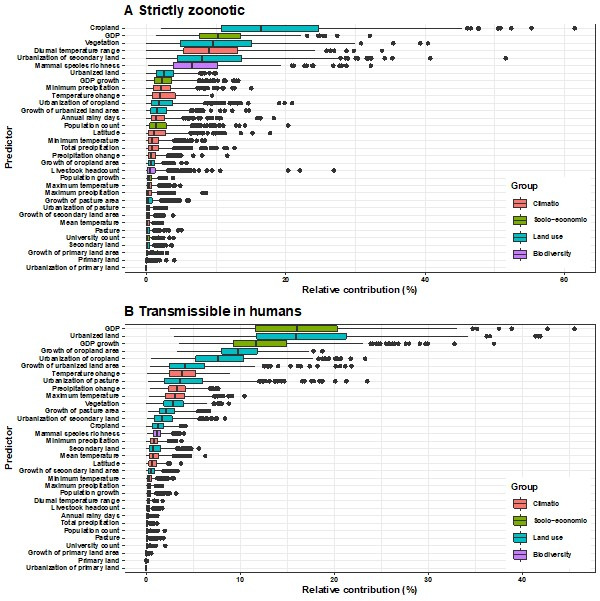
Background: The variation in the pathogen type as well as the spatial heterogeneity of predictors make the generality of any associations with pathogen discovery debatable. Our previous work confirmed that the association of a group of predictors differed across different types of RNA viruses, yet there have been no previous comparisons of the specific predictors for RNA virus discovery in different regions. The aim of the current study was to close the gap by investigating whether predictors of discovery rates within three regions-the United States, China, and Africa-differ from one another and from those at the global level.
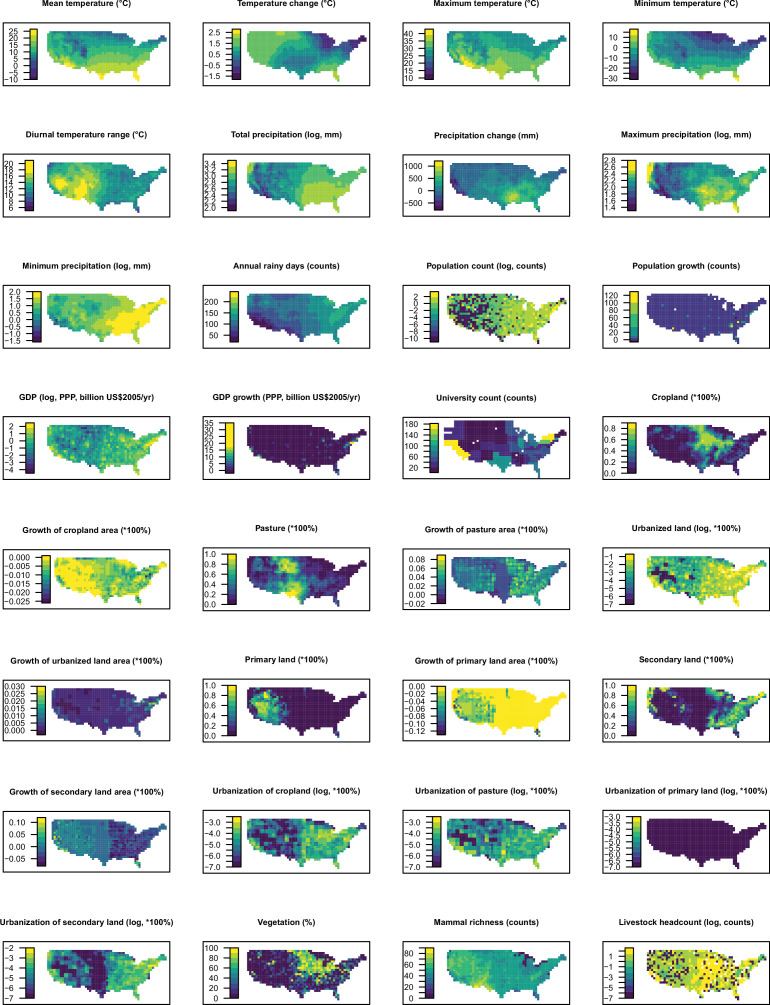
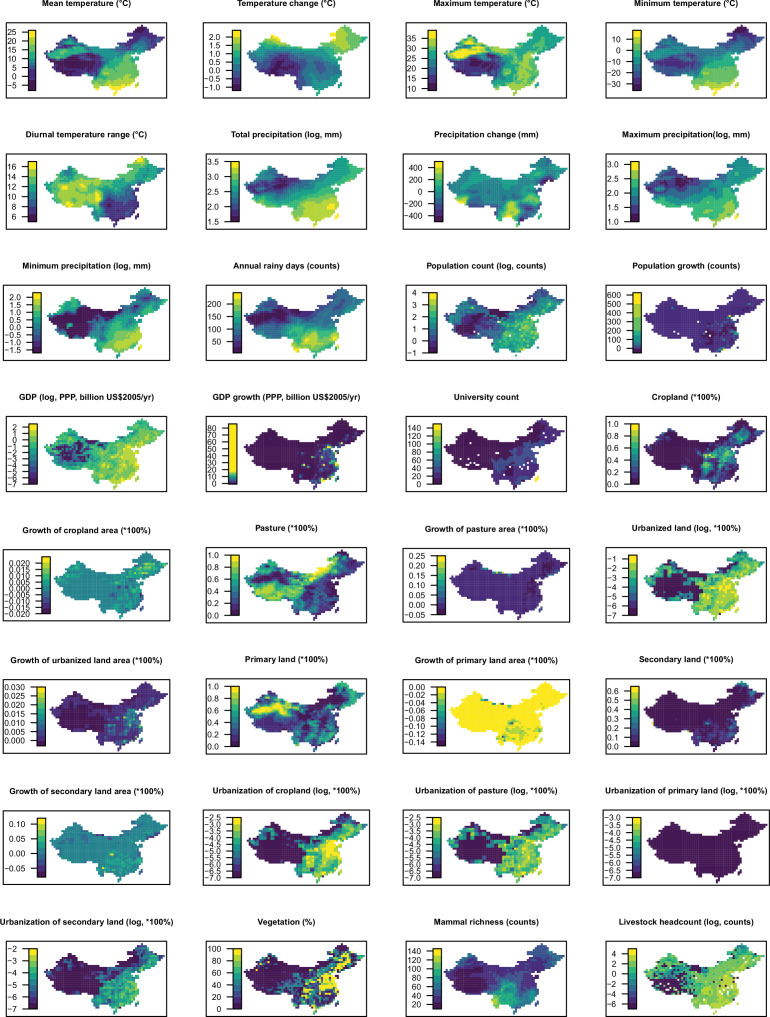
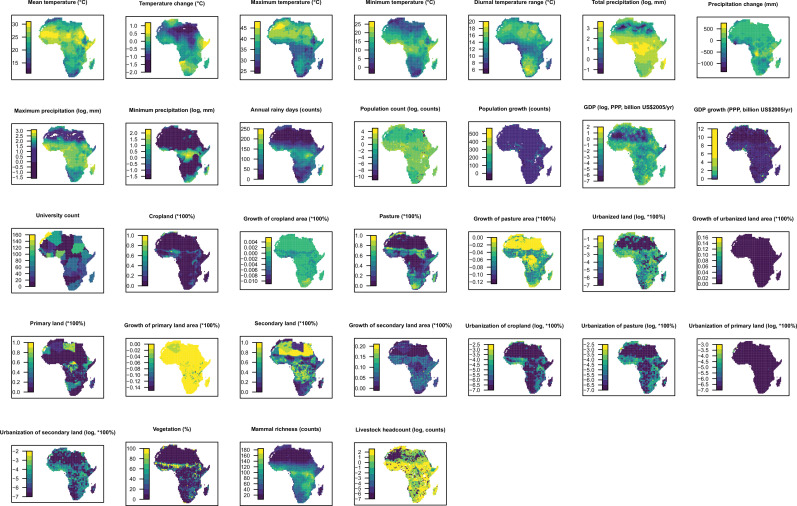
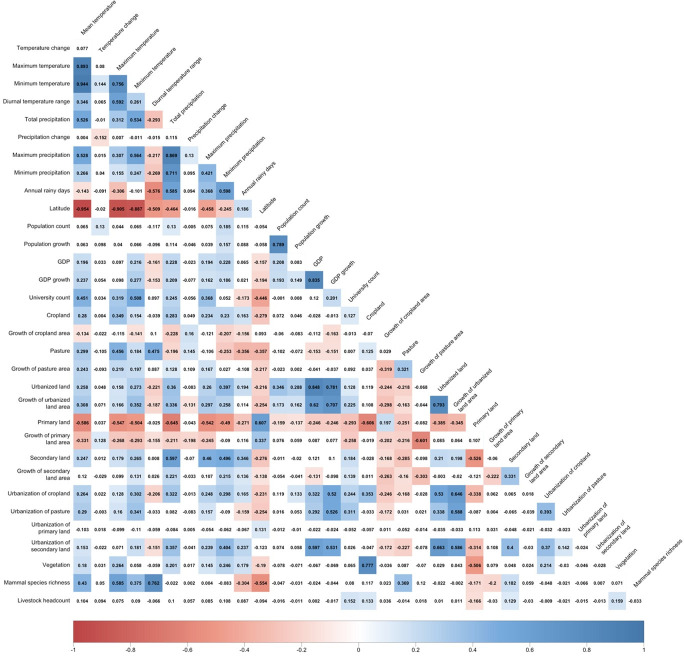
Methods: Based on a comprehensive list of human-infective RNA viruses, we collated published data on first discovery of each species in each region. We used a Poisson boosted regression tree (BRT) model to examine the relationship between virus discovery and 33 predictors representing climate, socio-economics, land use, and biodiversity across each region separately. The discovery probability in three regions in 2010-2019 was mapped using the fitted models and historical predictors.
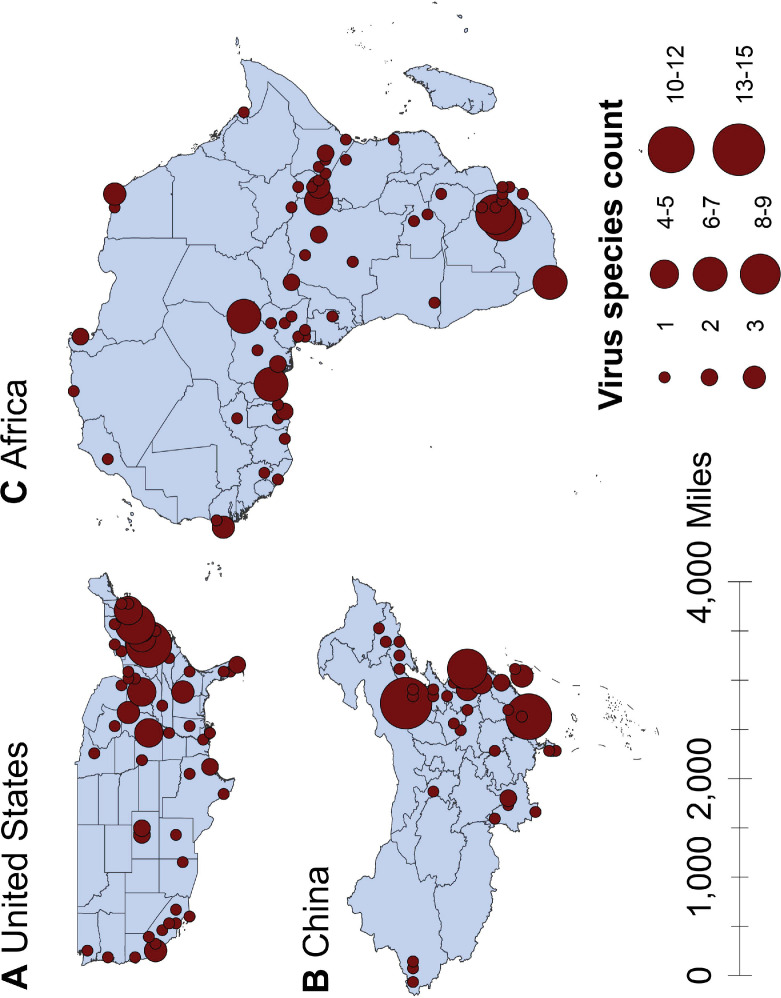
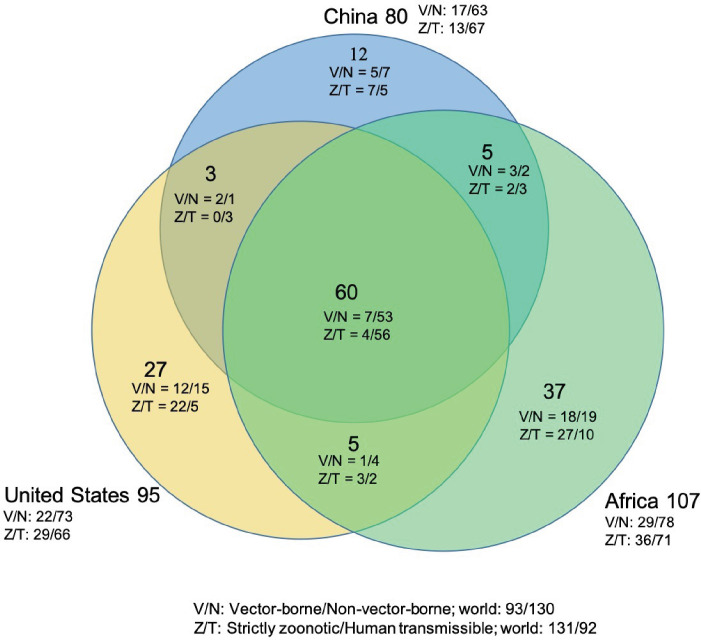
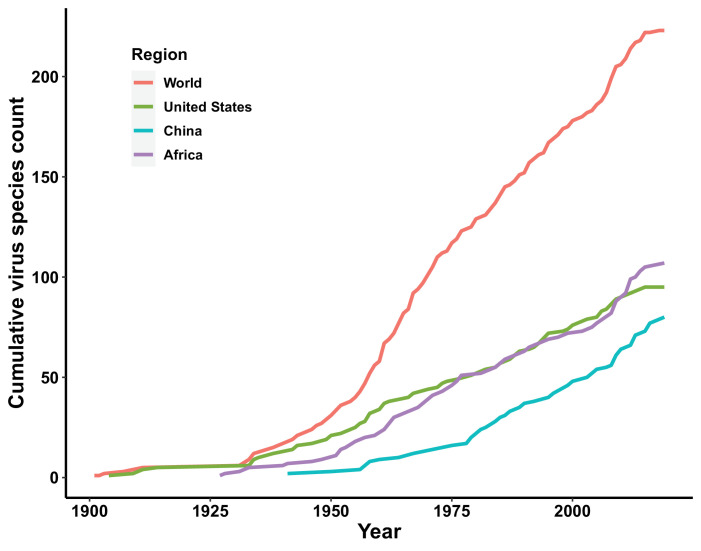
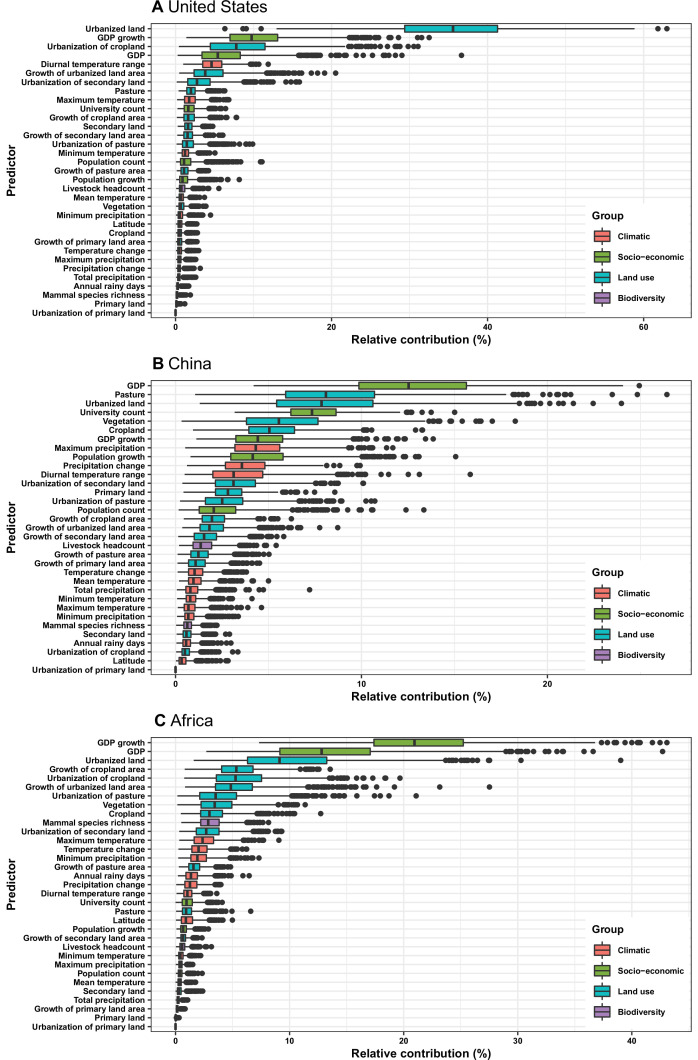
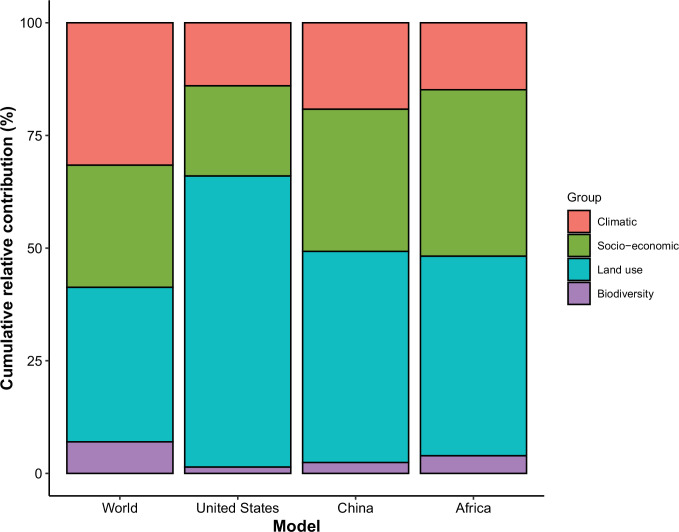
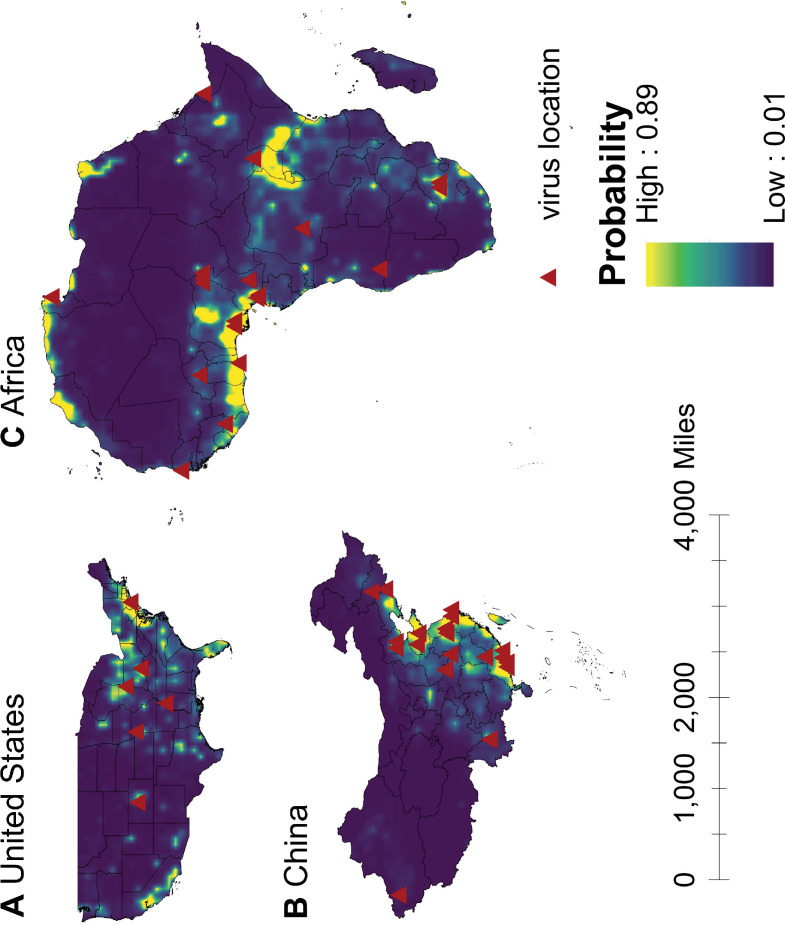
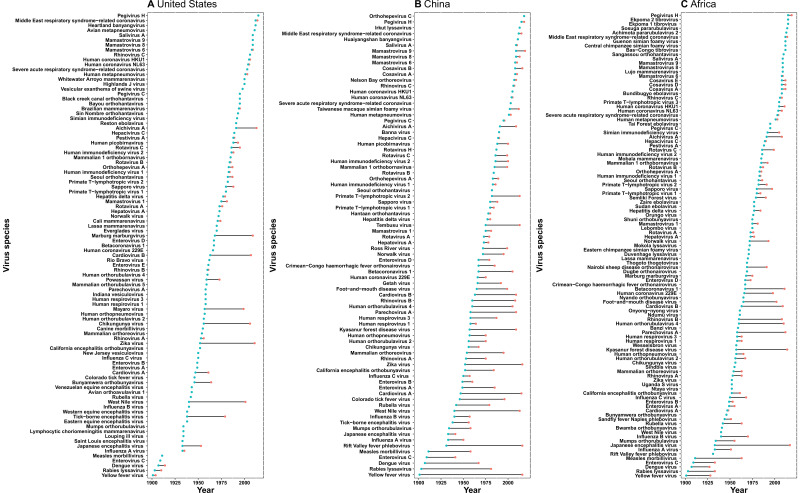
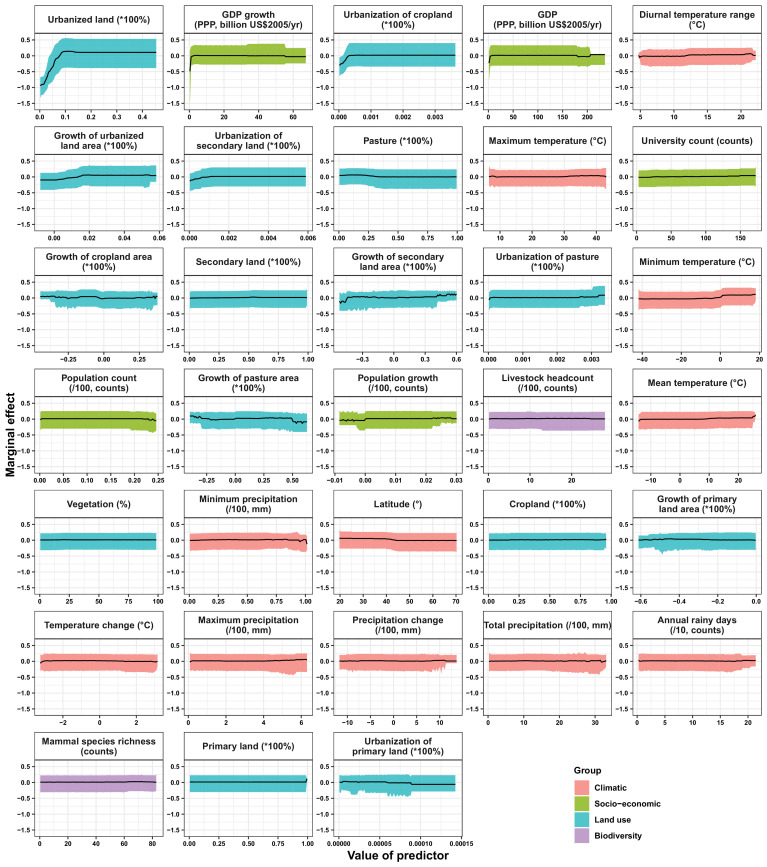
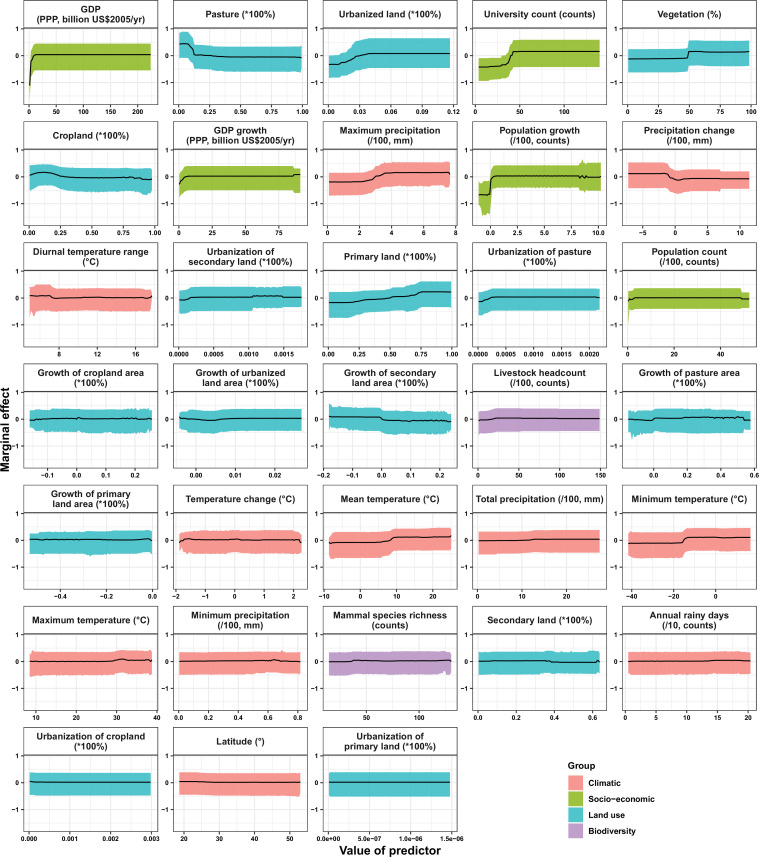
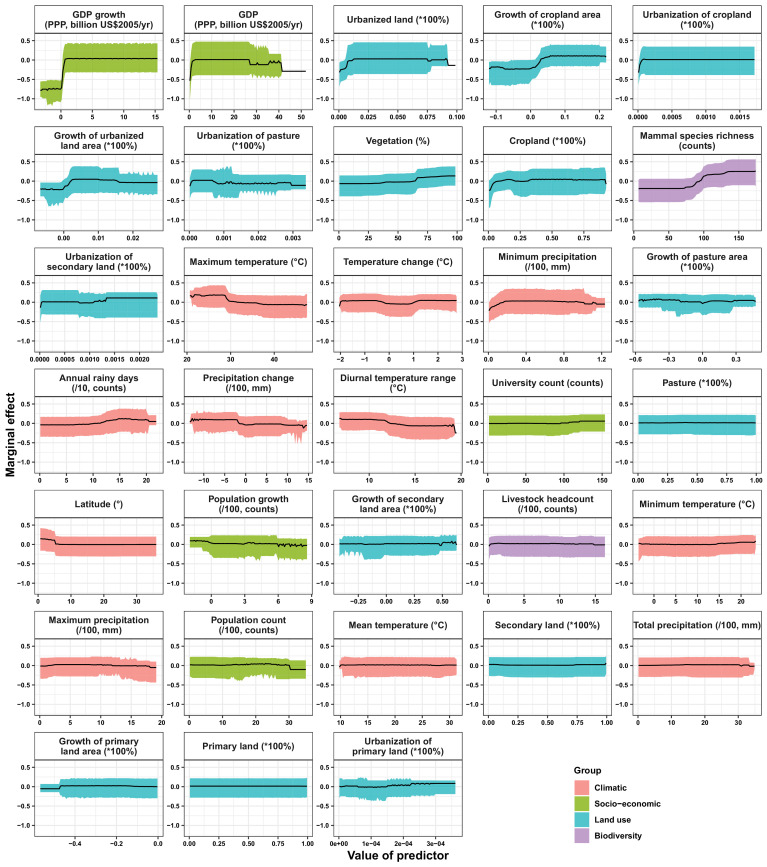
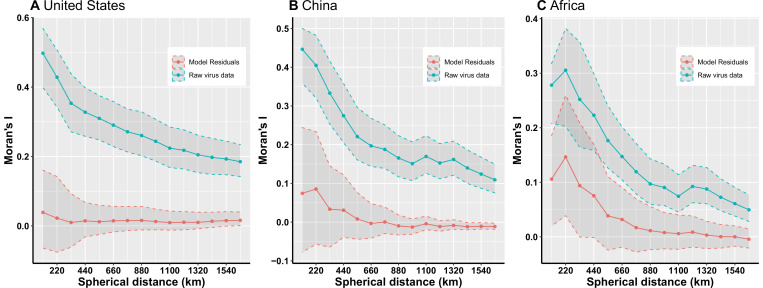
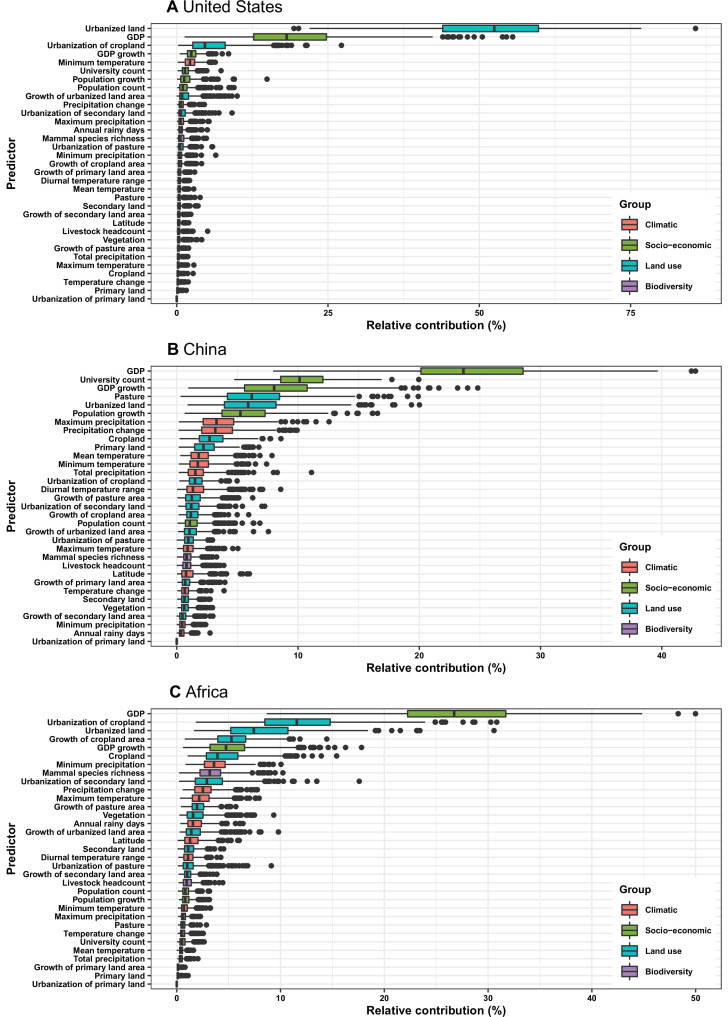
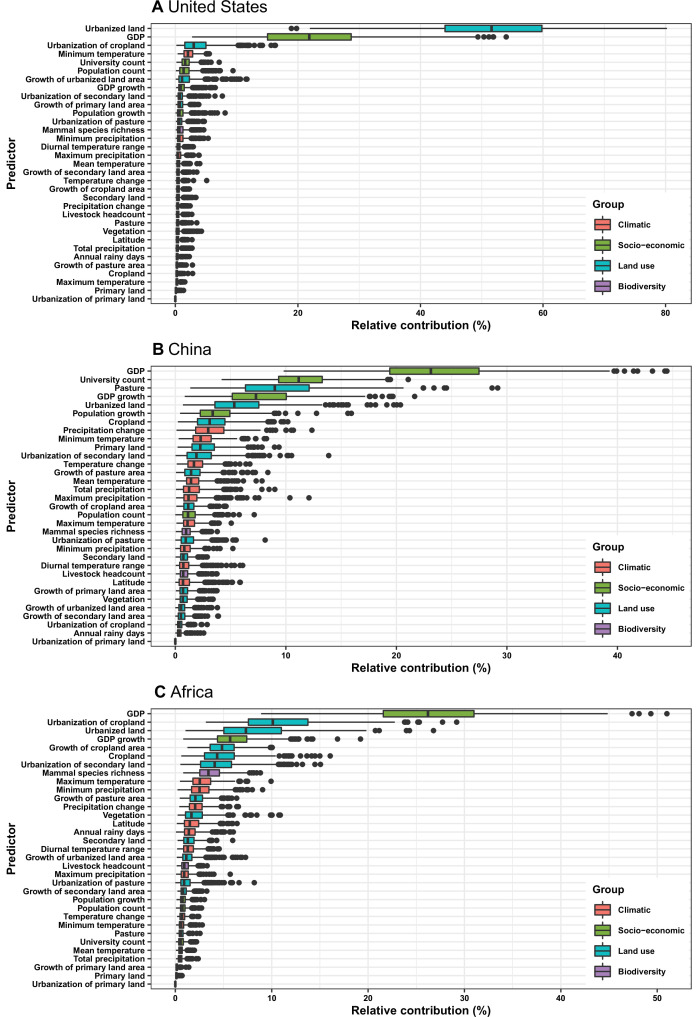
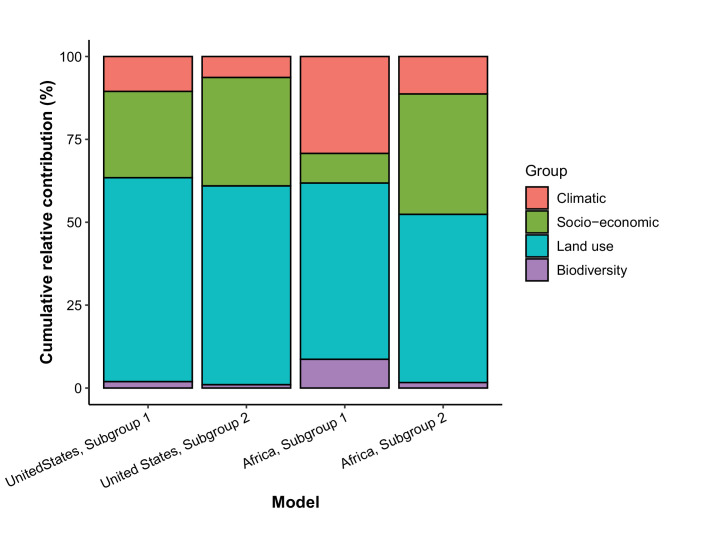
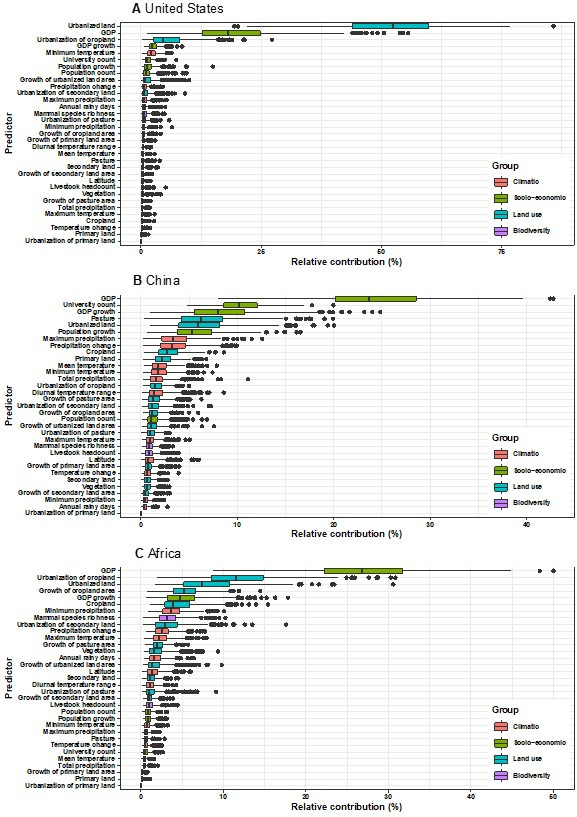
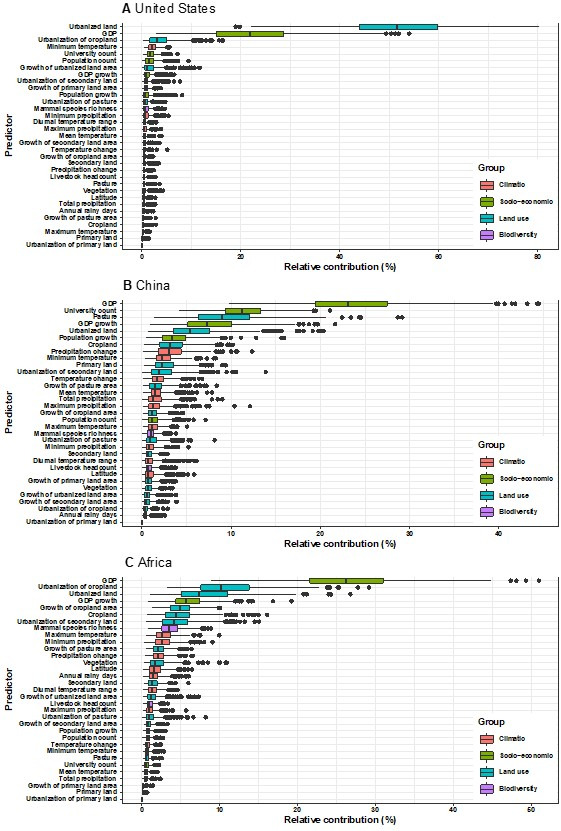
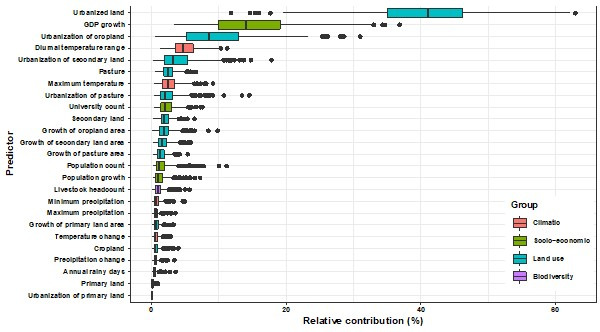
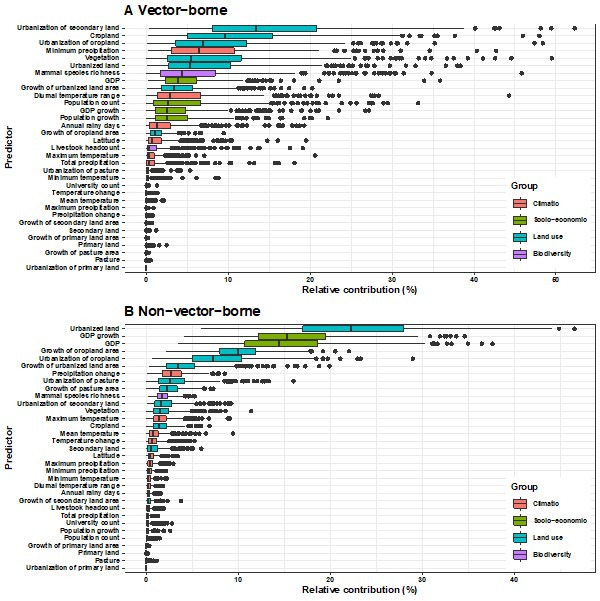
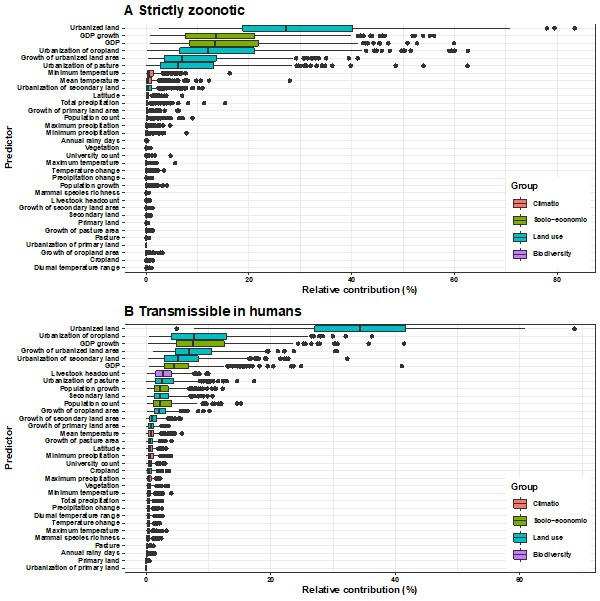
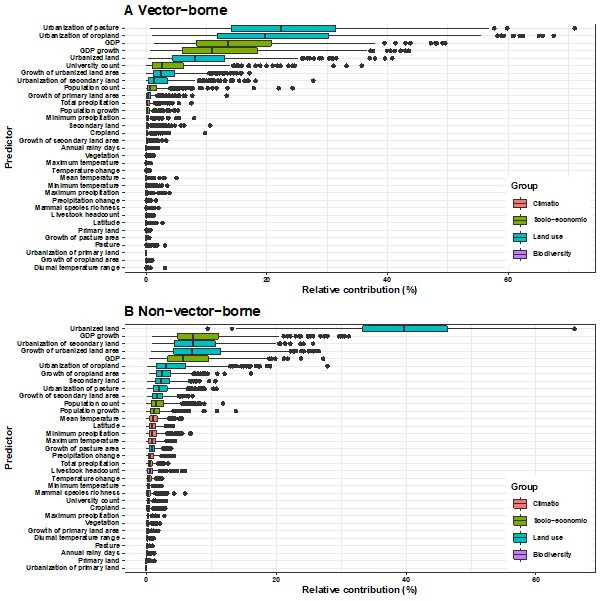
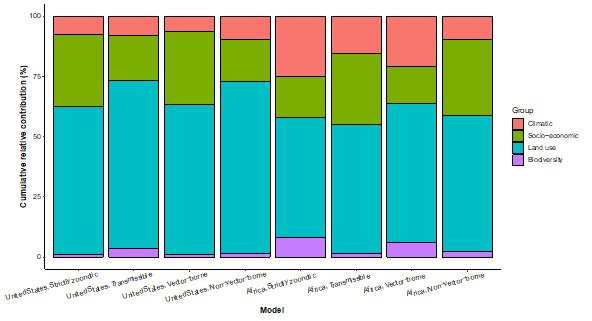
Results: The numbers of human-infective virus species discovered in the United States, China, and Africa up to 2019 were 95, 80, and 107 respectively, with China lagging behind the other two regions. In each region, discoveries were clustered in hotspots. BRT modelling suggested that in all three regions RNA virus discovery was better predicted by land use and socio-economic variables than climatic variables and biodiversity, although the relative importance of these predictors varied by region. Map of virus discovery probability in 2010-2019 indicated several new hotspots outside historical high-risk areas. Most new virus species since 2010 in each region (6/6 in the United States, 19/19 in China, 12/19 in Africa) were discovered in high-risk areas as predicted by our model.
Conclusions: The drivers of spatiotemporal variation in virus discovery rates vary in different regions of the world. Within regions virus discovery is driven mainly by land-use and socio-economic variables; climate and biodiversity variables are consistently less important predictors than at a global scale. Potential new discovery hotspots in 2010-2019 are identified. Results from the study could guide active surveillance for new human-infective viruses in local high-risk areas.
Funding: FFZ is funded by the Darwin Trust of Edinburgh (https://darwintrust.bio.ed.ac.uk/). MEJW has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement No. 874735 (VEO) (https://www.veo-europe.eu/).
Keywords: ecology; emerging virus; epidemiology; global health; machine learning; risk factor; viruses.
© 2022, Zhang et al.
Conflict of interest statement
FZ, MC, CG, MW No competing interests declared
Figures
References
-
- Abroug F, Slim A, Ouanes-Besbes L, Hadj Kacem M-A, Dachraoui F, Ouanes I, Lu X, Tao Y, Paden C, Caidi H, Miao C, Al-Hajri MM, Zorraga M, Ghaouar W, BenSalah A, Gerber SI, World Health Organization Global Outbreak Alert and Response Network Middle East Respiratory Syndrome Coronavirus International Investigation Team Family cluster of Middle East respiratory syndrome coronavirus infections, Tunisia, 2013. Emerging Infectious Diseases. 2014;20:1527–1530. doi: 10.3201/eid2009.140378. - DOI - PMC - PubMed
-
- Achong BG, Mansell PW, Epstein MA. A new human virus in cultures from A nasopharyngeal carcinoma. The Journal of Pathology. 1971;103:P18. - PubMed
-
- Albariño CG, Shoemaker T, Khristova ML, Wamala JF, Muyembe JJ, Balinandi S, Tumusiime A, Campbell S, Cannon D, Gibbons A, Bergeron E, Bird B, Dodd K, Spiropoulou C, Erickson BR, Guerrero L, Knust B, Nichol ST, Rollin PE, Ströher U. Genomic analysis of filoviruses associated with four viral hemorrhagic fever outbreaks in Uganda and the Democratic Republic of the Congo in 2012. Virology. 2013;442:97–100. doi: 10.1016/j.virol.2013.04.014. - DOI - PMC - PubMed
-
- Albariño CG, Foltzer M, Towner JS, Rowe LA, Campbell S, Jaramillo CM, Bird BH, Reeder DM, Vodzak ME, Rota P, Metcalfe MG, Spiropoulou CF, Knust B, Vincent JP, Frace MA, Nichol ST, Rollin PE, Ströher U. Novel paramyxovirus associated with severe acute febrile disease, South Sudan and Uganda, 2012. Emerging Infectious Diseases. 2014;20:211–216. doi: 10.3201/eid2002.131620. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
LinkOut - more resources
Full Text Sources
Miscellaneous
