

Evolving strategies for management of desmoid tumor
- PMID: 35670122
- PMCID: PMC9546183
- DOI: 10.1002/cncr.34332
Evolving strategies for management of desmoid tumor
Abstract
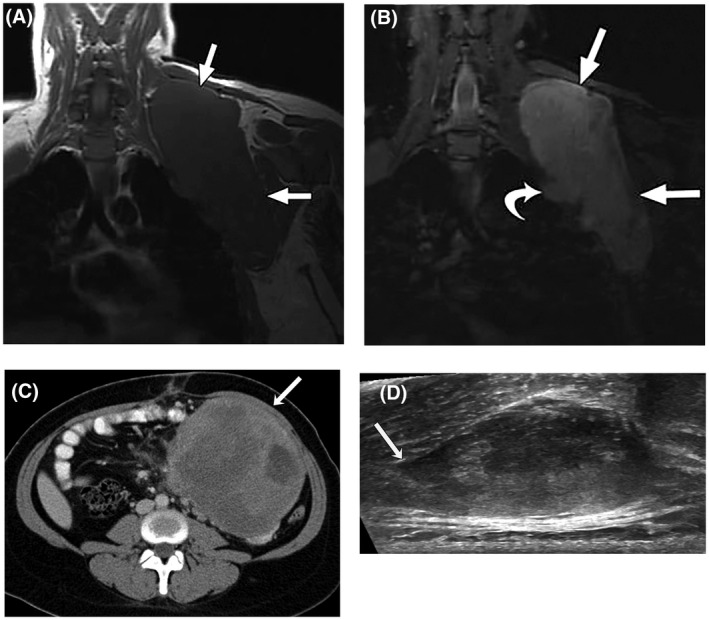
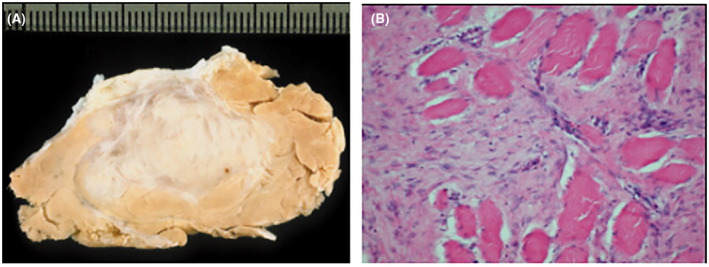
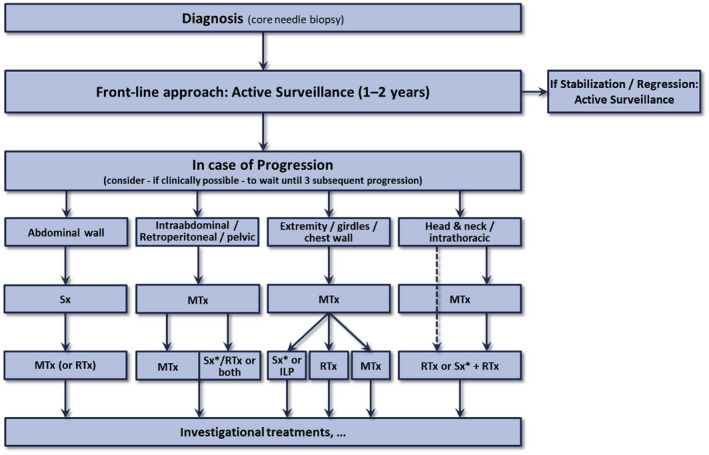
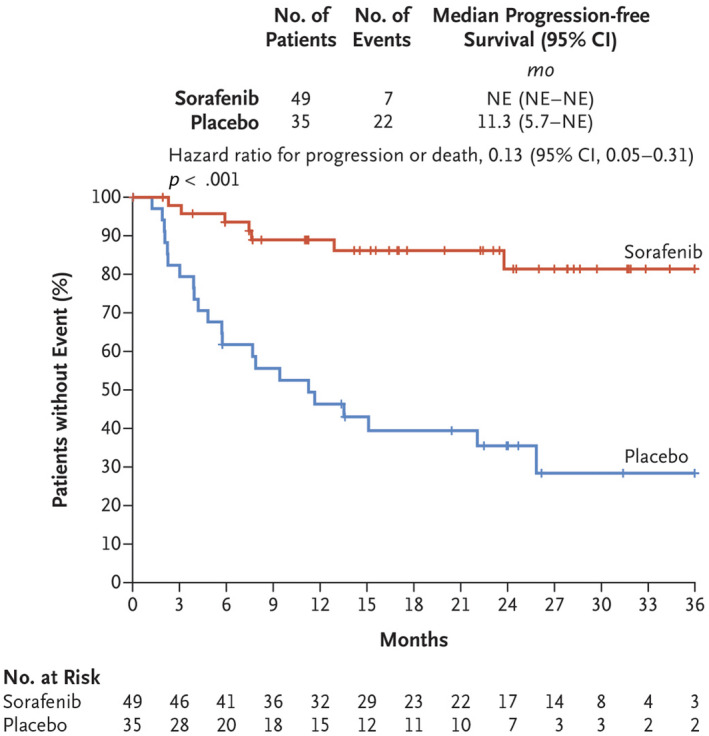
Desmoid tumors (DTs) are rare soft tissue mesenchymal neoplasms that may be associated with impairments, disfigurement, morbidity, and (rarely) mortality. DT disease course can be unpredictable. Most DTs are sporadic, harboring somatic mutations in the gene that encodes for β-catenin, whereas DTs occurring in patients with familial adenomatous polyposis have germline mutations in the APC gene, which encodes for a protein regulator of β-catenin. Pathology review by an expert soft tissue pathologist is critical in making a diagnosis. Magnetic resonance imaging is preferred for most anatomic locations. Surgery, once the standard of care for initial treatment of DT, is associated with a significant risk of recurrence as well as avoidable morbidity because spontaneous regressions are known to occur without treatment. Consequently, active surveillance in conjunction with pain management is now recommended for most patients. Systemic medical treatment of DT has evolved beyond the use of hormone therapy, which is no longer routinely recommended. Current options for medical management include tyrosine kinase inhibitors as well as more conventional cytotoxic chemotherapy (e.g., anthracycline-based or methotrexate-based regimens). A newer class of agents, γ-secretase inhibitors, appears promising, including in patients who fail other therapies, but confirmation in Phase 3 trials is needed. In summary, DTs present challenges to physicians in diagnosis and prognosis, as well as in determining treatment initiation, type, duration, and sequence. Accordingly, evaluation by a multidisciplinary team with expertise in DT and patient-tailored management are essential. As management strategies continue to evolve, further studies will help clarify these issues and optimize outcomes for patients.
Keywords: active surveillance; antineoplastic agents; desmoid tumor; fibromatosis, aggressive; radiotherapy; tyrosine kinase inhibitors; γ-secretase inhibitors.
© 2022 The Authors. Cancer published by Wiley Periodicals LLC on behalf of American Cancer Society.
Conflict of interest statement
Richard F. Riedel reports institutional clinical research support from Aadi, AROG, Ayala, BioAtla, Daiichi‐Sankyo, Deciphera, GlaxoSmithKline, Ignyta, Inhibrx, Immune Design, Karyopharm, Lilly, NanoCarrier, Novartis, Oncternal, Philogen, Plexxikon, Rain Therapeutics, Roche, SpringWorks Therapeutics, Threshold, Tracon, and Trillium; personal/advisory fees from Aadi, Bayer, Blueprint, Daiichi‐Sankyo, Deciphera, EISAI, EMD Serono, Janssen, Lilly, Ignyta, NanoCarrier, and SpringWorks Therapeutics; and ownership in Limbguard, LLC (spouse), all outside the submitted work. Mark Agulnik reports research funding from Exelixis and personal/advisory fees from Aadi, Bayer, Adaptimmune, Regeneron, AstraZeneca, Bristol‐Myers Squibb, and Deciphera, all outside the submitted work.
Figures
References
-
- World Health Organization Classification of Tumours Editorial Board . WHO Classification of Tumours: Soft Tissue and Bone Tumours. Soft Tissue and Bone Tumours. Volume 3. IARC Press; 2020.
-
- Constantinidou A, Scurr M, Judson I, Litchman C. Clinical presentation of desmoid tumor. In: Litchman C, ed. Desmoid Tumor. Springer Science; 2012:5‐16.
-
- Kasper B, Baumgarten C, Garcia J, et al. An update on the management of sporadic desmoid‐type fibromatosis: a European Consensus Initiative between Sarcoma PAtients EuroNet (SPAEN) and European Organization for Research and Treatment of Cancer (EORTC)/Soft Tissue and Bone Sarcoma Group (STBSG). Ann Oncol. 2017;28(10):2399‐2408. - PMC - PubMed
-
- Shinagare AB, Ramaiya NH, Jagannathan JP, et al. A to Z of desmoid tumors. AJR Am J Roentgenol. 2011;197(6):W1008‐W1014. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
