

Schwann cell nodal membrane disruption triggers bystander axonal degeneration in a Guillain-Barré syndrome mouse model
- PMID: 35671105
- PMCID: PMC9282931
- DOI: 10.1172/JCI158524
Schwann cell nodal membrane disruption triggers bystander axonal degeneration in a Guillain-Barré syndrome mouse model
Abstract
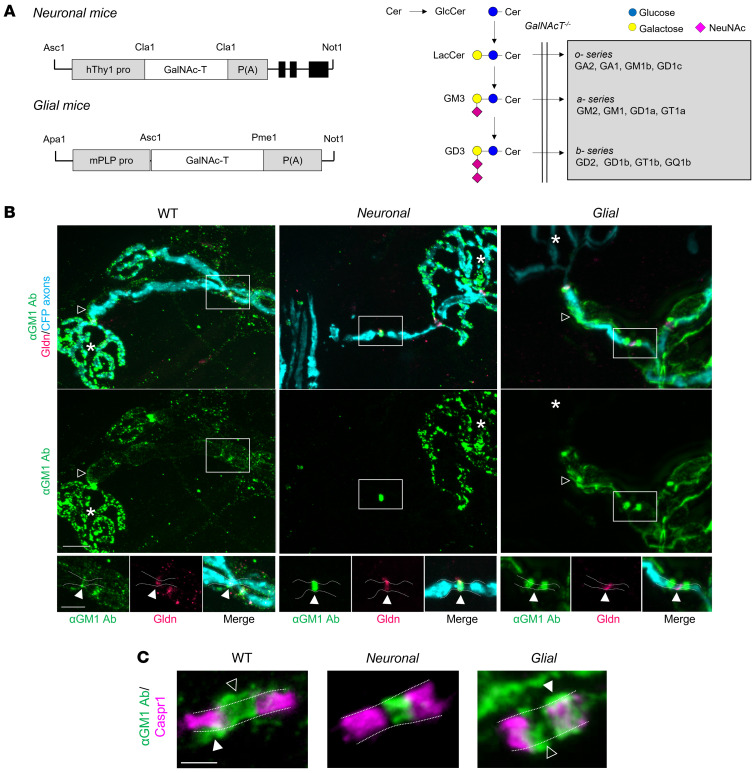
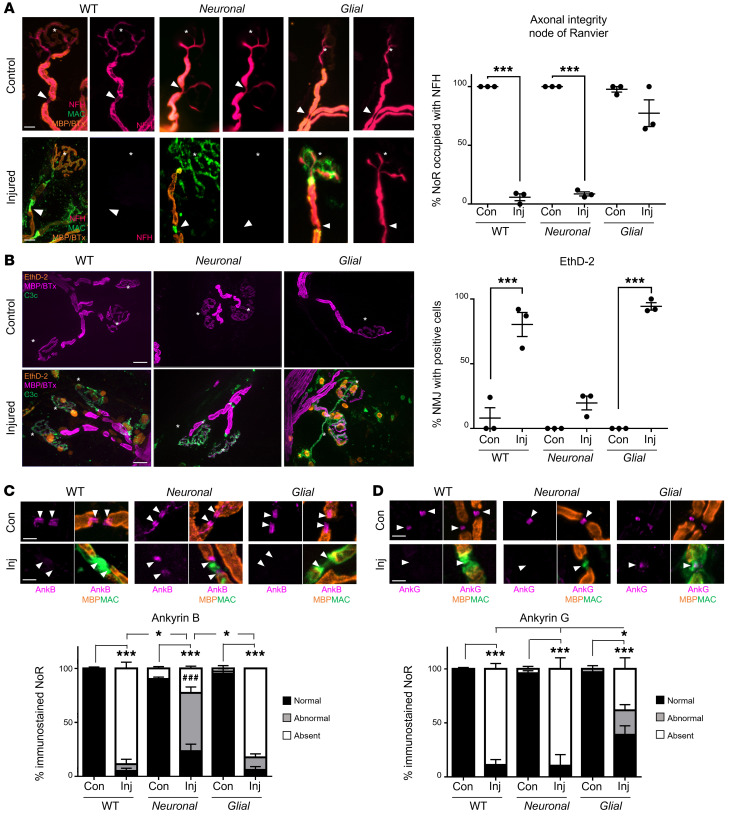
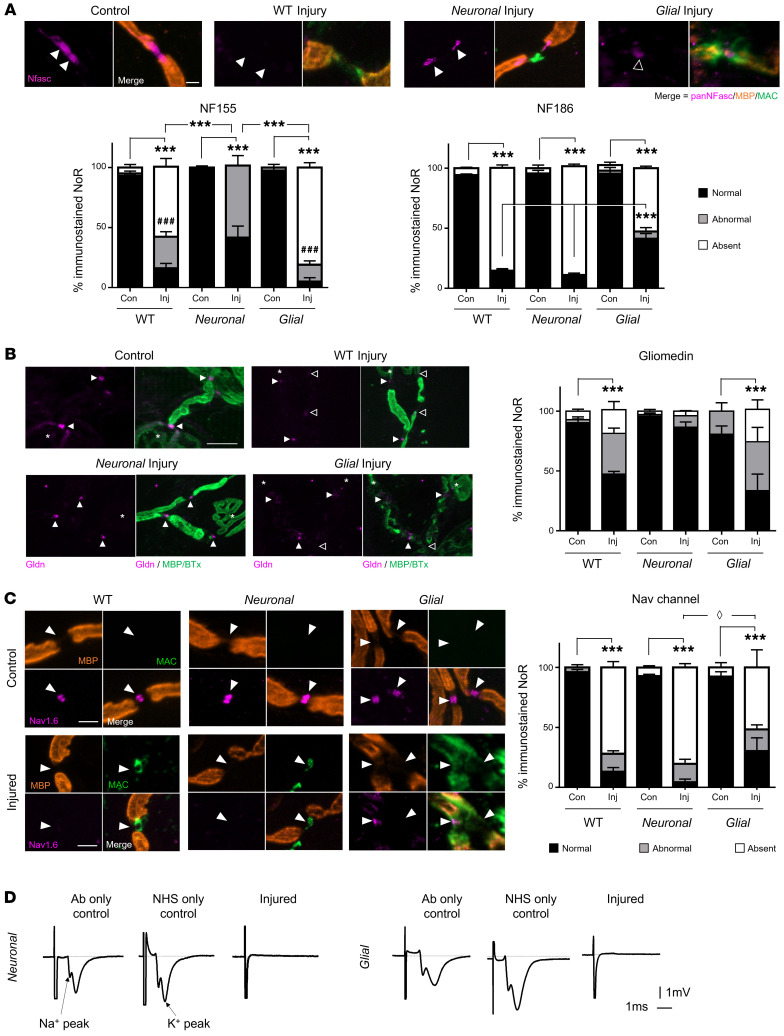
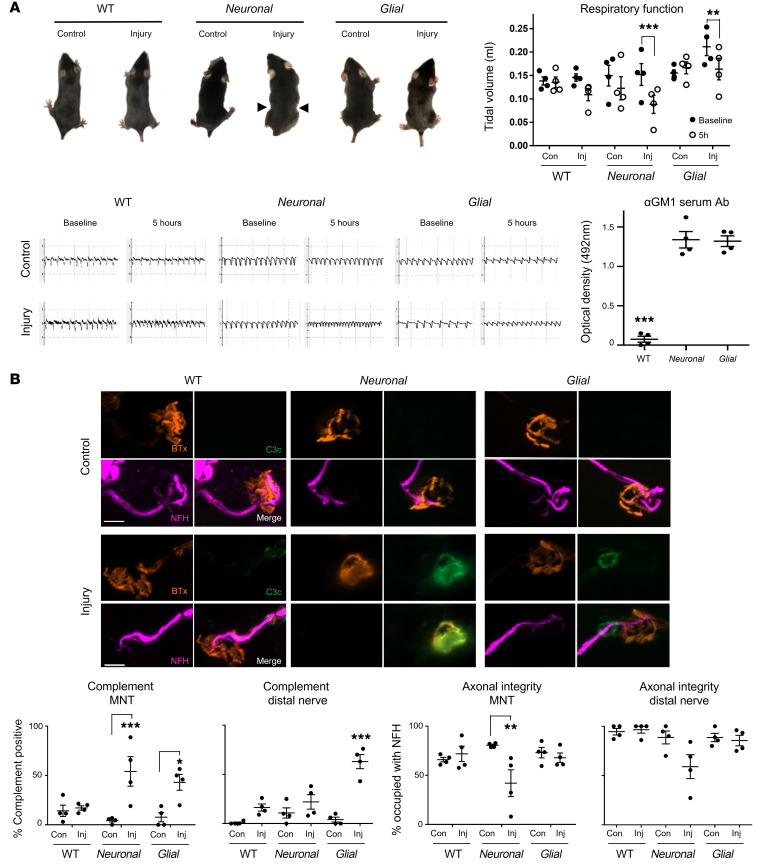
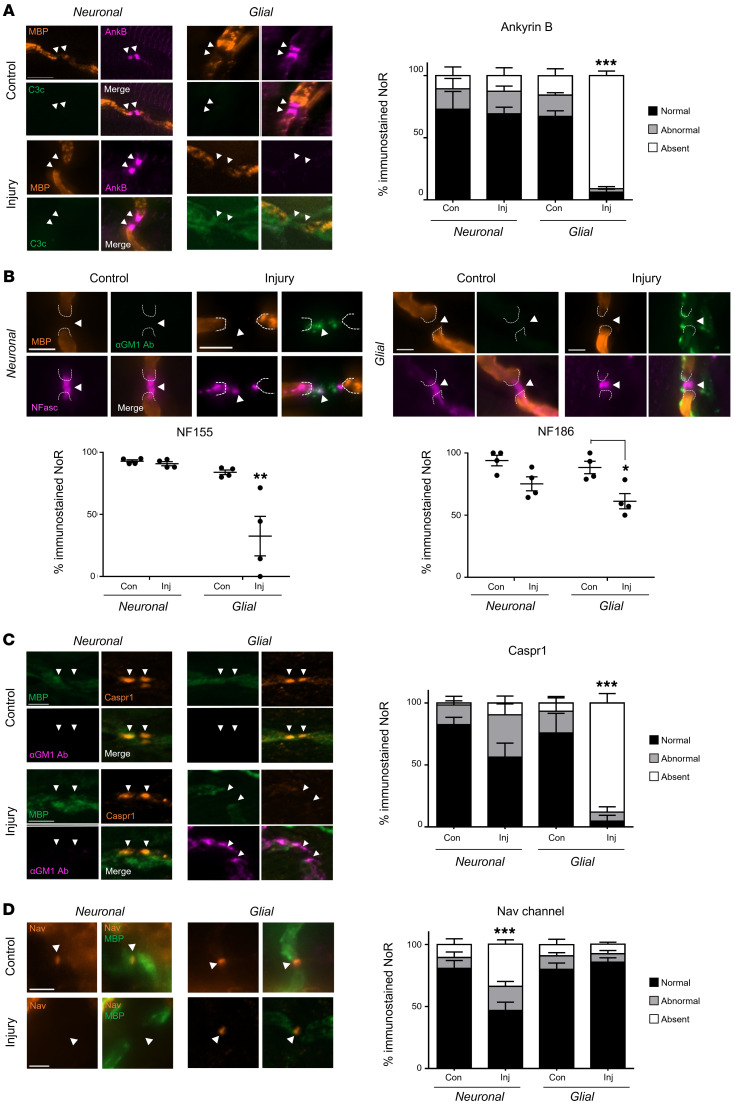
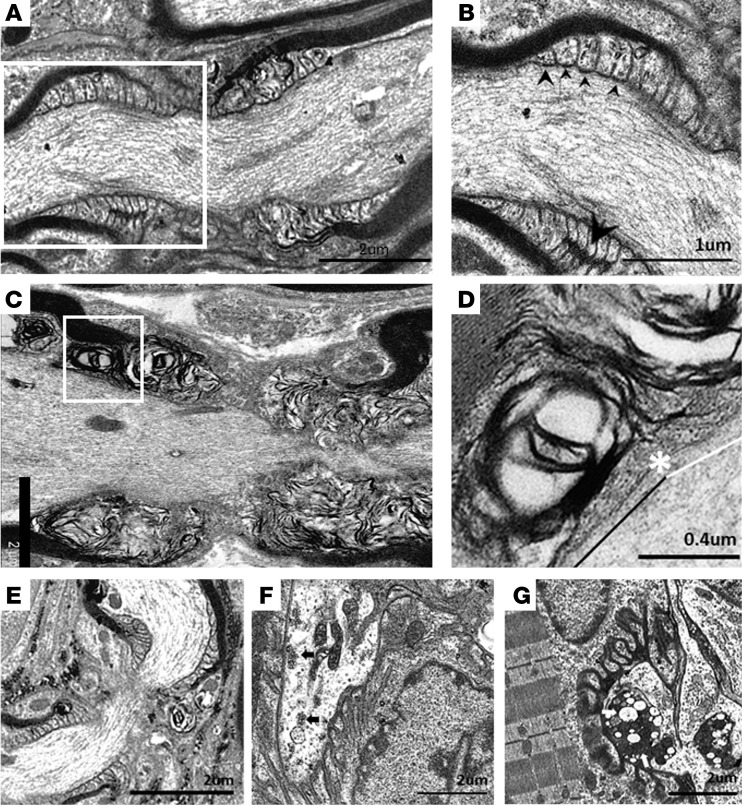
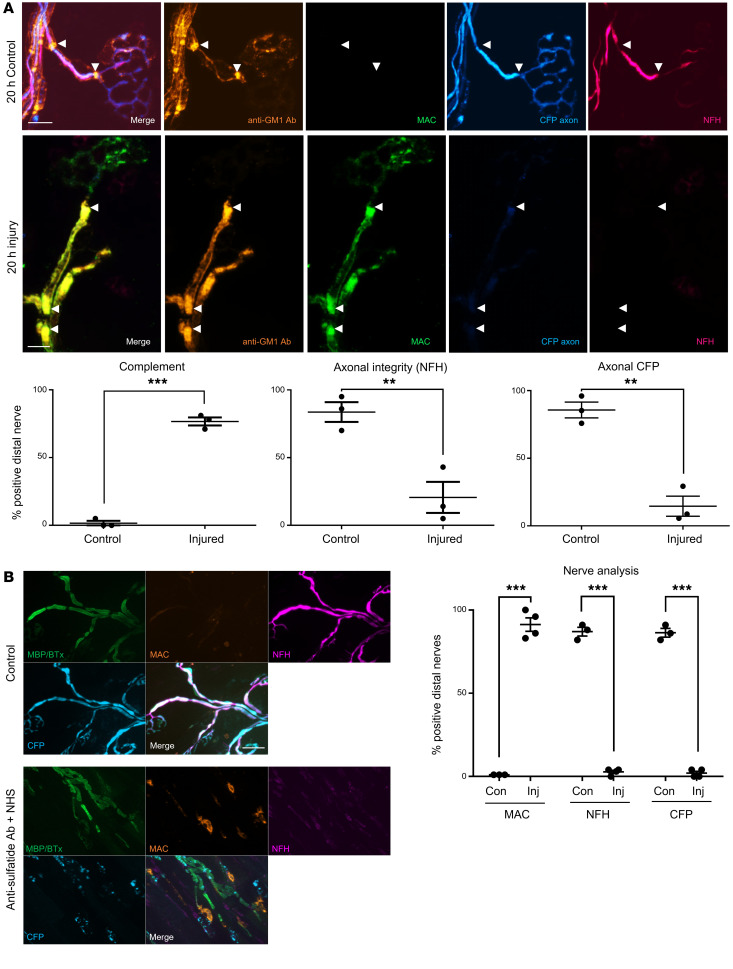
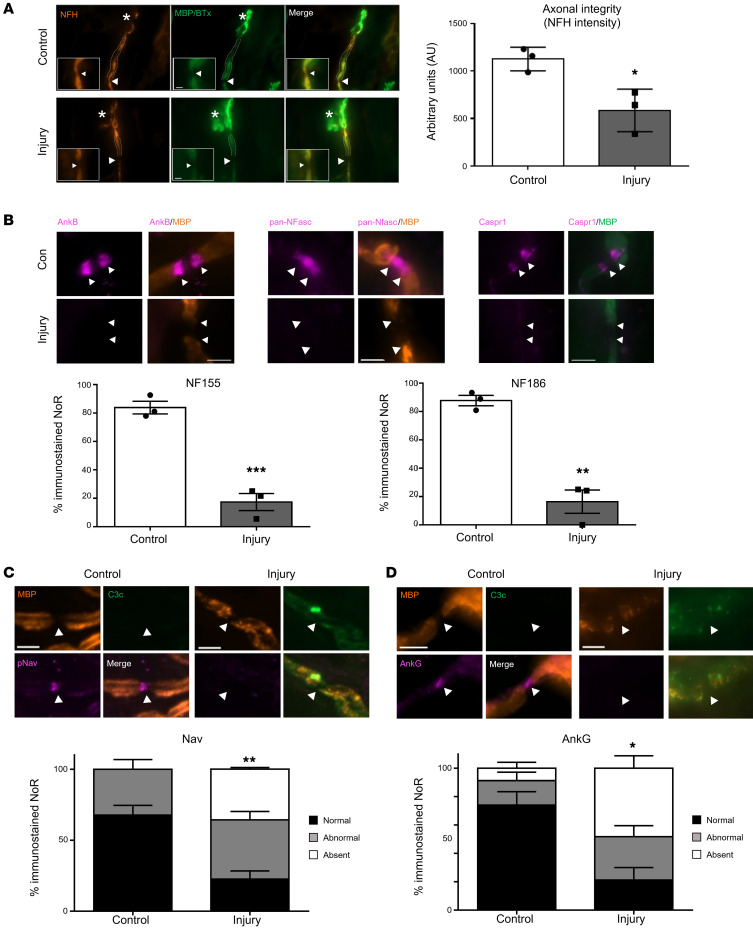
In Guillain-Barré syndrome (GBS), both axonal and demyelinating variants can be mediated by complement-fixing anti-GM1 ganglioside autoantibodies that target peripheral nerve axonal and Schwann cell (SC) membranes, respectively. Critically, the extent of axonal degeneration in both variants dictates long-term outcome. The differing pathomechanisms underlying direct axonal injury and the secondary bystander axonal degeneration following SC injury are unresolved. To investigate this, we generated glycosyltransferase-disrupted transgenic mice that express GM1 ganglioside either exclusively in neurons [GalNAcT-/--Tg(neuronal)] or glia [GalNAcT-/--Tg(glial)], thereby allowing anti-GM1 antibodies to solely target GM1 in either axonal or SC membranes, respectively. Myelinated-axon integrity in distal motor nerves was studied in transgenic mice exposed to anti-GM1 antibody and complement in ex vivo and in vivo injury paradigms. Axonal targeting induced catastrophic acute axonal disruption, as expected. When mice with GM1 in SC membranes were targeted, acute disruption of perisynaptic glia and SC membranes at nodes of Ranvier (NoRs) occurred. Following glial injury, axonal disruption at NoRs also developed subacutely, progressing to secondary axonal degeneration. These models differentiate the distinctly different axonopathic pathways under axonal and glial membrane targeting conditions, and provide insights into primary and secondary axonal injury, currently a major unsolved area in GBS research.
Keywords: Autoimmunity; Demyelinating disorders; Neuroscience.
Conflict of interest statement
Figures
Similar articles
-
Complement inhibition prevents glial nodal membrane injury in a GM1 antibody-mediated mouse model.Brain Commun. 2022 Nov 23;4(6):fcac306. doi: 10.1093/braincomms/fcac306. eCollection 2022. Brain Commun. 2022. PMID: 36523267 Free PMC article.
-
C1q-targeted inhibition of the classical complement pathway prevents injury in a novel mouse model of acute motor axonal neuropathy.Acta Neuropathol Commun. 2016 Mar 2;4:23. doi: 10.1186/s40478-016-0291-x. Acta Neuropathol Commun. 2016. PMID: 26936605 Free PMC article.
-
Axolemmal nanoruptures arising from paranodal membrane injury induce secondary axon degeneration in murine Guillain-Barré syndrome.J Peripher Nerv Syst. 2023 Mar;28(1):17-31. doi: 10.1111/jns.12532. Epub 2023 Feb 12. J Peripher Nerv Syst. 2023. PMID: 36710500 Free PMC article.
-
The immunobiology of Guillain-Barré syndromes.J Peripher Nerv Syst. 2005 Jun;10(2):94-112. doi: 10.1111/j.1085-9489.2005.0010202.x. J Peripher Nerv Syst. 2005. PMID: 15958123 Review.
-
Ganglioside mimicry as a cause of Guillain-Barré syndrome.Curr Opin Neurol. 2005 Oct;18(5):557-61. doi: 10.1097/01.wco.0000174604.42272.2d. Curr Opin Neurol. 2005. PMID: 16155440 Review.
Cited by
-
Guillain-Barré syndrome.Nat Rev Dis Primers. 2024 Dec 19;10(1):97. doi: 10.1038/s41572-024-00580-4. Nat Rev Dis Primers. 2024. PMID: 39702645
-
Neuromuscular disease: 2023 update.Free Neuropathol. 2023 Feb 27;4:2. doi: 10.17879/freeneuropathology-2023-4682. eCollection 2023 Jan. Free Neuropathol. 2023. PMID: 37283936 Free PMC article.
-
Real time imaging of intra-axonal calcium flux in an explant mouse model of axonal Guillain-Barré syndrome.Exp Neurol. 2022 Sep;355:114127. doi: 10.1016/j.expneurol.2022.114127. Epub 2022 May 29. Exp Neurol. 2022. PMID: 35640716 Free PMC article.
-
Guillain-Barré syndrome: a comprehensive review.Eur J Neurol. 2024 Aug;31(8):e16365. doi: 10.1111/ene.16365. Epub 2024 May 30. Eur J Neurol. 2024. PMID: 38813755 Free PMC article. Review.
-
Complement inhibition prevents glial nodal membrane injury in a GM1 antibody-mediated mouse model.Brain Commun. 2022 Nov 23;4(6):fcac306. doi: 10.1093/braincomms/fcac306. eCollection 2022. Brain Commun. 2022. PMID: 36523267 Free PMC article.
References
-
- Feasby TE, et al. An acute axonal form of Guillain-Barré polyneuropathy. Brain. 1986;109(pt 6):1115–1126. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
