

Language models can learn complex molecular distributions
- PMID: 35672310
- PMCID: PMC9174447
- DOI: 10.1038/s41467-022-30839-x
Language models can learn complex molecular distributions
Abstract
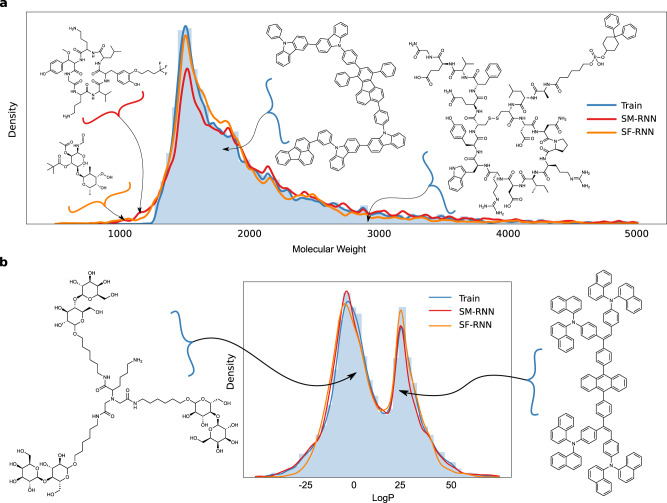
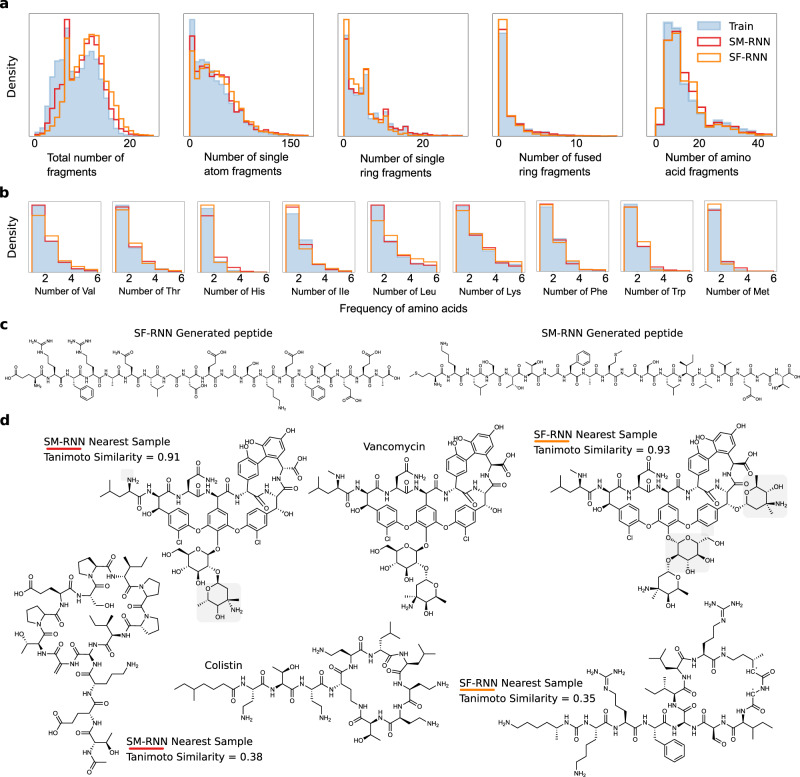
Deep generative models of molecules have grown immensely in popularity, trained on relevant datasets, these models are used to search through chemical space. The downstream utility of generative models for the inverse design of novel functional compounds, depends on their ability to learn a training distribution of molecules. The most simple example is a language model that takes the form of a recurrent neural network and generates molecules using a string representation. Since their initial use, subsequent work has shown that language models are very capable, in particular, recent research has demonstrated their utility in the low data regime. In this work, we investigate the capacity of simple language models to learn more complex distributions of molecules. For this purpose, we introduce several challenging generative modeling tasks by compiling larger, more complex distributions of molecules and we evaluate the ability of language models on each task. The results demonstrate that language models are powerful generative models, capable of adeptly learning complex molecular distributions. Language models can accurately generate: distributions of the highest scoring penalized LogP molecules in ZINC15, multi-modal molecular distributions as well as the largest molecules in PubChem. The results highlight the limitations of some of the most popular and recent graph generative models- many of which cannot scale to these molecular distributions.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
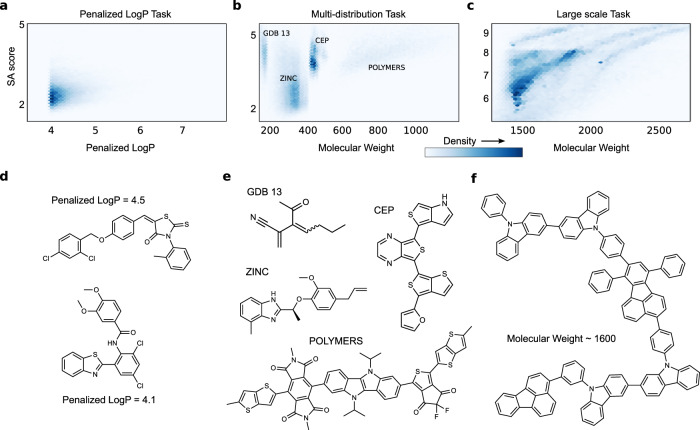
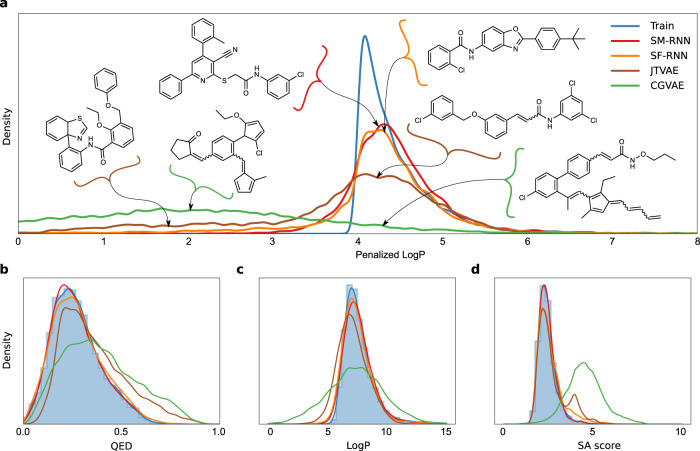
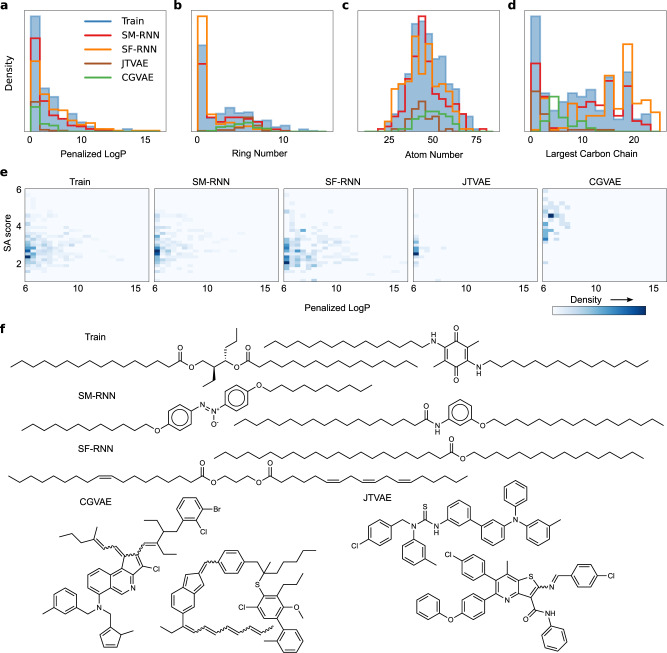
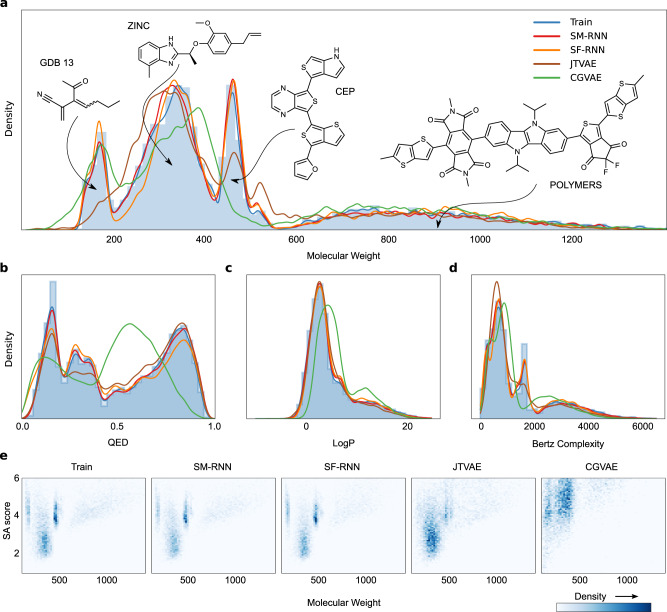
References
-
- Sutskever, I., Martens, J. & Hinton, G. E. Generating text with recurrent neural networks. In International Conference on Machine Learning (2011).
-
- Weininger D. Smiles, a chemical language and information system. 1. introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988;28:31. doi: 10.1021/ci00057a005. - DOI
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
