

QMugs, quantum mechanical properties of drug-like molecules
- PMID: 35672335
- PMCID: PMC9174255
- DOI: 10.1038/s41597-022-01390-7
QMugs, quantum mechanical properties of drug-like molecules
Abstract
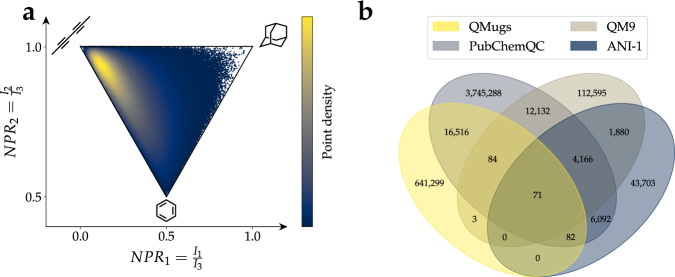
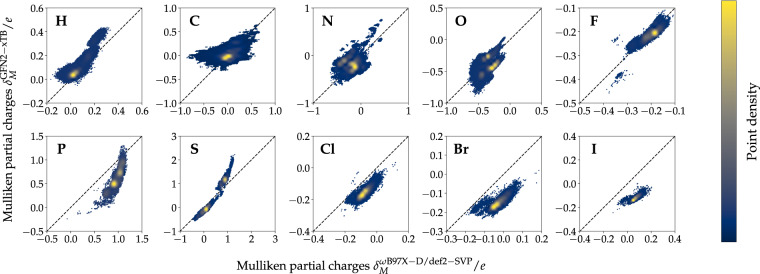
Machine learning approaches in drug discovery, as well as in other areas of the chemical sciences, benefit from curated datasets of physical molecular properties. However, there currently is a lack of data collections featuring large bioactive molecules alongside first-principle quantum chemical information. The open-access QMugs (Quantum-Mechanical Properties of Drug-like Molecules) dataset fills this void. The QMugs collection comprises quantum mechanical properties of more than 665 k biologically and pharmacologically relevant molecules extracted from the ChEMBL database, totaling ~2 M conformers. QMugs contains optimized molecular geometries and thermodynamic data obtained via the semi-empirical method GFN2-xTB. Atomic and molecular properties are provided on both the GFN2-xTB and on the density-functional levels of theory (DFT, ωB97X-D/def2-SVP). QMugs features molecules of significantly larger size than previously-reported collections and comprises their respective quantum mechanical wave functions, including DFT density and orbital matrices. This dataset is intended to facilitate the development of models that learn from molecular data on different levels of theory while also providing insight into the corresponding relationships between molecular structure and biological activity.
© 2022. The Author(s).
Conflict of interest statement
G.S. is a cofounder of inSili.com LLC, Zurich, and a consultant to the pharmaceutical industry.
Figures





References
-
- Schmidt J, Marques MR, Botti S, Marques MA. Recent advances and applications of machine learning in solid-state materials science. Npj Comput. Mater. 2019;5:83. doi: 10.1038/s41524-019-0221-0. - DOI
-
- Satorras, V. G., Hoogeboom, E. & Welling, M. E(n) equivariant graph neural networks. In International Conference on Machine Learning, 9323–9332 (PMLR, 2021).
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
