Single-cell RNA-sequencing of peripheral blood mononuclear cells reveals widespread, context-specific gene expression regulation upon pathogenic exposure
- PMID: 35672358
- PMCID: PMC9174272
- DOI: 10.1038/s41467-022-30893-5
Single-cell RNA-sequencing of peripheral blood mononuclear cells reveals widespread, context-specific gene expression regulation upon pathogenic exposure
Abstract
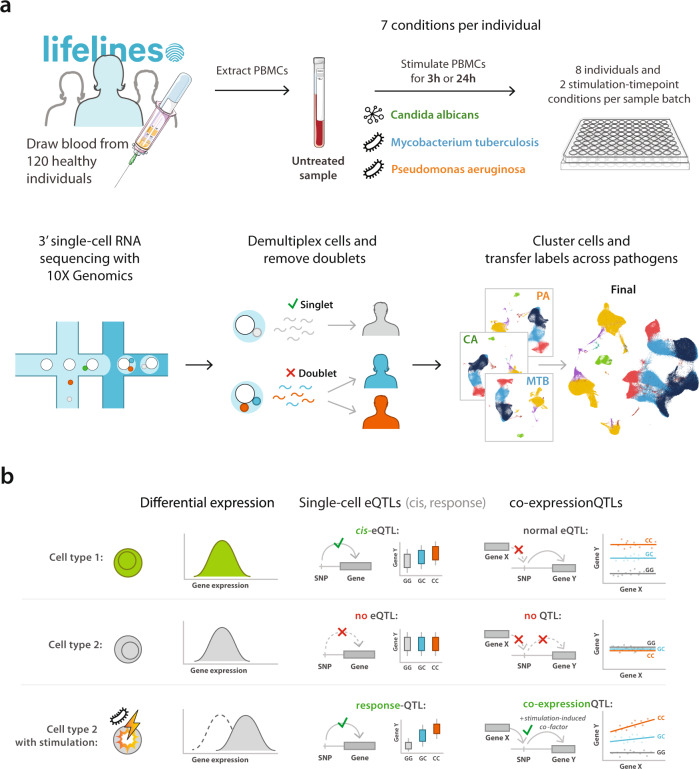
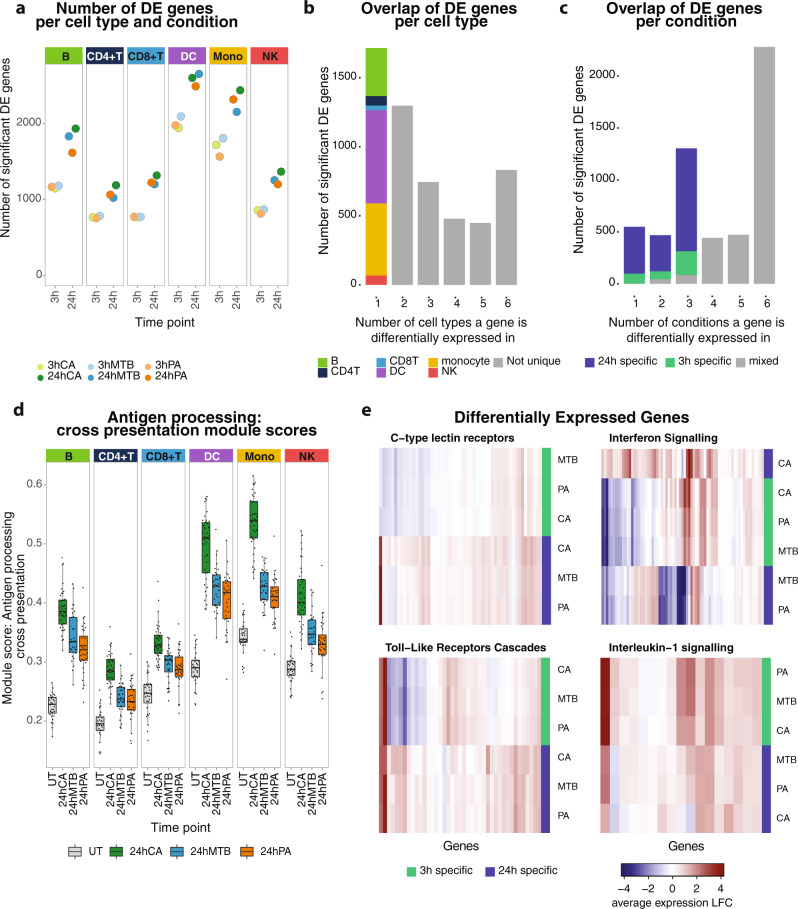
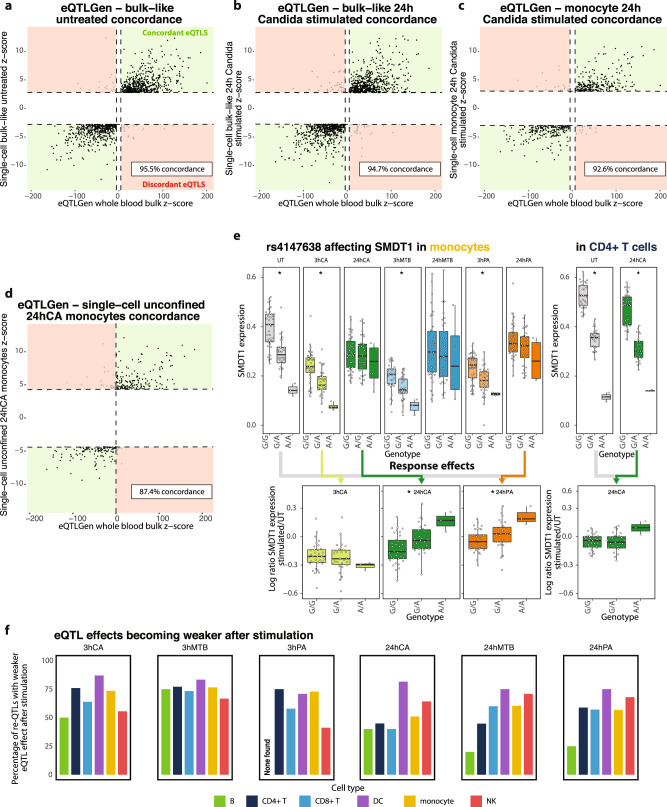
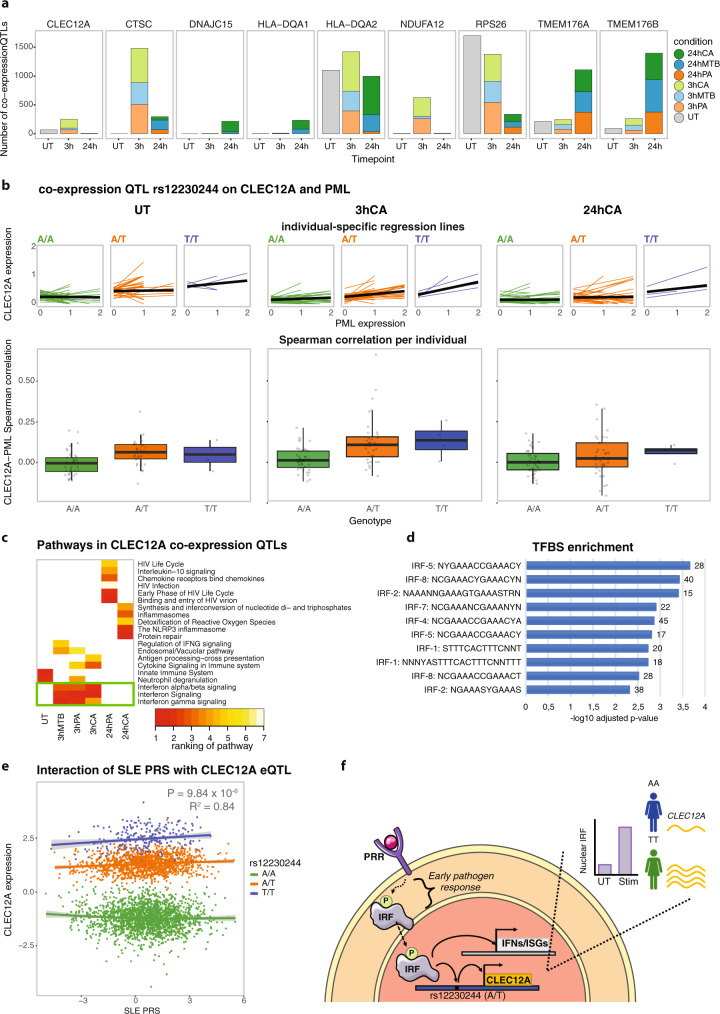
The host's gene expression and gene regulatory response to pathogen exposure can be influenced by a combination of the host's genetic background, the type of and exposure time to pathogens. Here we provide a detailed dissection of this using single-cell RNA-sequencing of 1.3M peripheral blood mononuclear cells from 120 individuals, longitudinally exposed to three different pathogens. These analyses indicate that cell-type-specificity is a more prominent factor than pathogen-specificity regarding contexts that affect how genetics influences gene expression (i.e., eQTL) and co-expression (i.e., co-expression QTL). In monocytes, the strongest responder to pathogen stimulations, 71.4% of the genetic variants whose effect on gene expression is influenced by pathogen exposure (i.e., response QTL) also affect the co-expression between genes. This indicates widespread, context-specific changes in gene expression level and its regulation that are driven by genetics. Pathway analysis on the CLEC12A gene that exemplifies cell-type-, exposure-time- and genetic-background-dependent co-expression interactions, shows enrichment of the interferon (IFN) pathway specifically at 3-h post-exposure in monocytes. Similar genetic background-dependent association between IFN activity and CLEC12A co-expression patterns is confirmed in systemic lupus erythematosus by in silico analysis, which implies that CLEC12A might be an IFN-regulated gene. Altogether, this study highlights the importance of context for gaining a better understanding of the mechanisms of gene regulation in health and disease.
© 2022. The Author(s).
Conflict of interest statement
C.J.Y. is a Scientific Advisory Board member for and hold equity in Related Sciences and ImmunAI, a consultant for and hold equity in Maze Therapeutics, and a consultant for TReX Bio. C.J.Y. has received research support from Chan Zuckerberg Initiative, Chan Zuckerberg Biohub, and Genentech. The remaining authors declare no competing interests.
Figures