

Decrypting a Cryptic Allosteric Pocket in H. pylori Glutamate Racemase
- PMID: 35673630
- PMCID: PMC9169614
- DOI: 10.1038/s42004-021-00605-z
Decrypting a Cryptic Allosteric Pocket in H. pylori Glutamate Racemase
Abstract
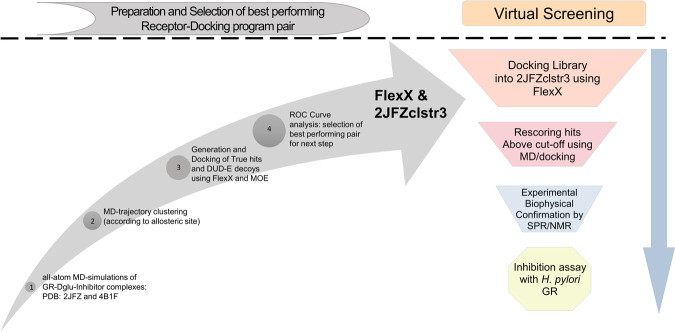
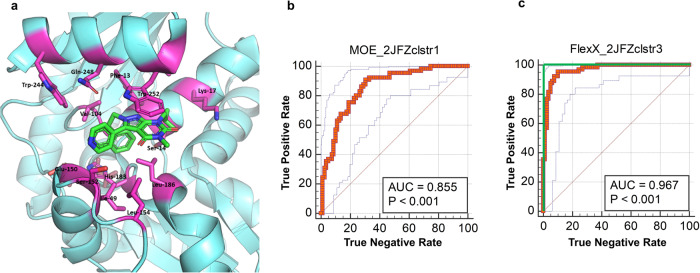
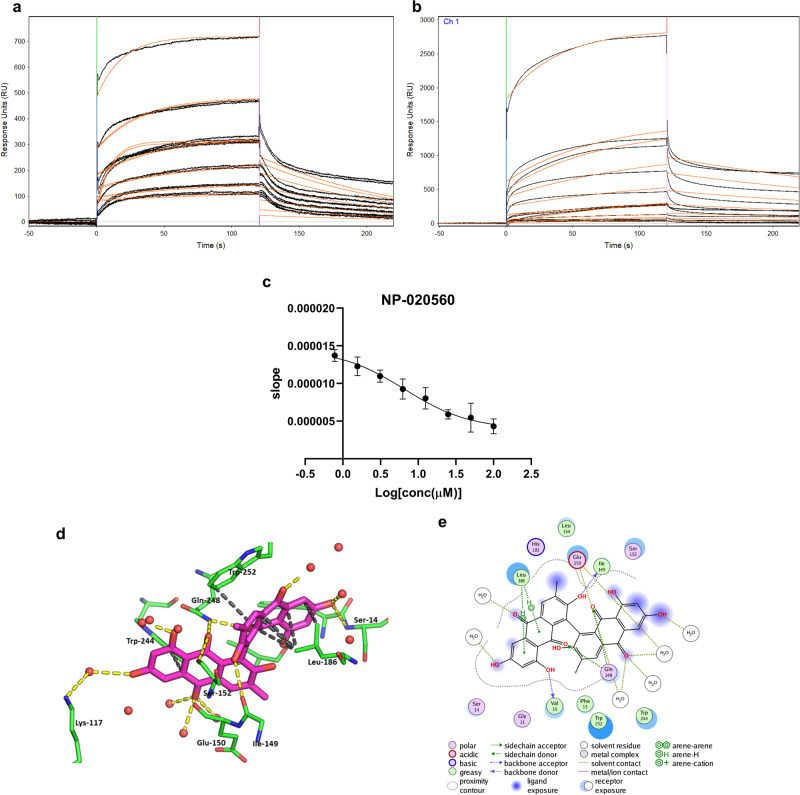
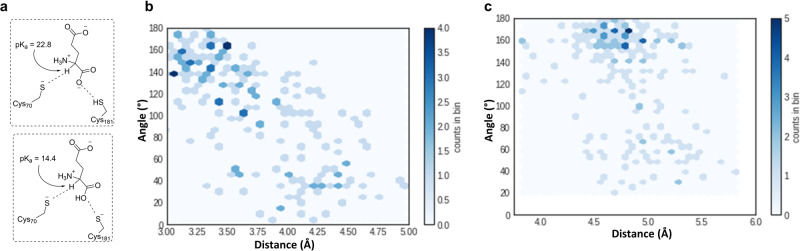
One of our greatest challenges in drug design is targeting cryptic allosteric pockets in enzyme targets. Drug leads that do bind to these cryptic pockets are often discovered during HTS campaigns, and the mechanisms of action are rarely understood. Nevertheless, it is often the case that the allosteric pocket provides the best option for drug development against a given target. In the current studies we present a successful way forward in rationally exploiting the cryptic allosteric pocket of H. pylori glutamate racemase, an essential enzyme in this pathogen's life cycle. A wide range of computational and experimental methods are employed in a workflow leading to the discovery of a series of natural product allosteric inhibitors which occupy the allosteric pocket of this essential racemase. The confluence of these studies reveals a fascinating source of the allosteric inhibition, which centers on the abolition of essential monomer-monomer coupled motion networks.
Conflict of interest statement
Competing interests The authors declare no competing interests.
Figures



Similar articles
-
Decrypting a cryptic allosteric pocket in H. pylori glutamate racemase.Commun Chem. 2021 Dec 10;4(1):172. doi: 10.1038/s42004-021-00605-z. Commun Chem. 2021. PMID: 36697800
-
Designing small molecules to target cryptic pockets yields both positive and negative allosteric modulators.PLoS One. 2017 Jun 1;12(6):e0178678. doi: 10.1371/journal.pone.0178678. eCollection 2017. PLoS One. 2017. PMID: 28570708 Free PMC article.
-
An Atomistic Understanding of Allosteric Inhibition of Glutamate Racemase: a Dampening of Native Activation Dynamics.ChemMedChem. 2020 Feb 17;15(4):376-384. doi: 10.1002/cmdc.201900642. Epub 2020 Jan 21. ChemMedChem. 2020. PMID: 31876113 Free PMC article.
-
Covalent allosteric modulation: An emerging strategy for GPCRs drug discovery.Eur J Med Chem. 2020 Nov 15;206:112690. doi: 10.1016/j.ejmech.2020.112690. Epub 2020 Aug 9. Eur J Med Chem. 2020. PMID: 32818870 Free PMC article. Review.
-
Allostery and Missense Mutations as Intermittently Linked Promising Aspects of Modern Computational Drug Discovery.J Mol Biol. 2022 Sep 15;434(17):167610. doi: 10.1016/j.jmb.2022.167610. Epub 2022 Apr 28. J Mol Biol. 2022. PMID: 35490897 Review.
References
Grants and funding
LinkOut - more resources
Full Text Sources
