

Deep Sequencing Analysis of Individual HIV-1 Proviruses Reveals Frequent Asymmetric Long Terminal Repeats
- PMID: 35674431
- PMCID: PMC9278108
- DOI: 10.1128/jvi.00122-22
Deep Sequencing Analysis of Individual HIV-1 Proviruses Reveals Frequent Asymmetric Long Terminal Repeats
Abstract
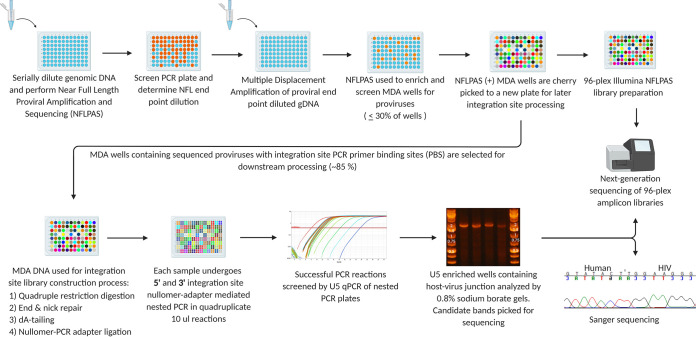
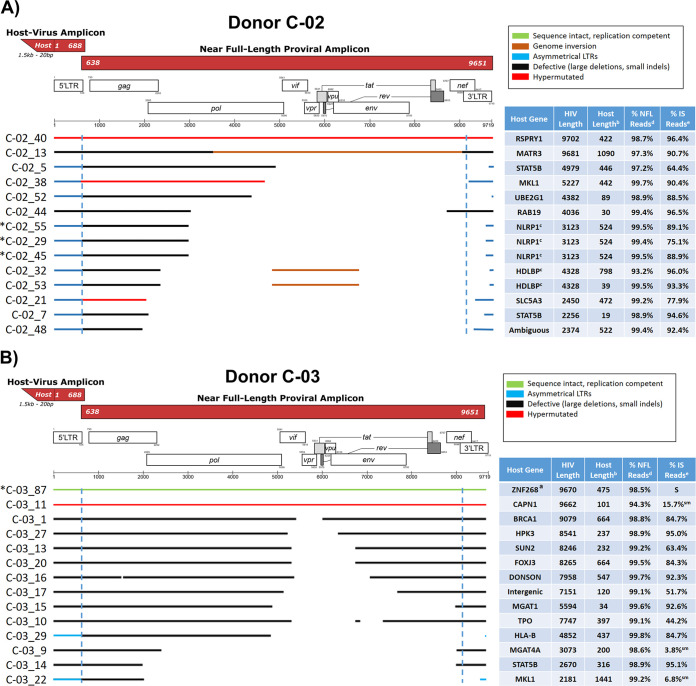
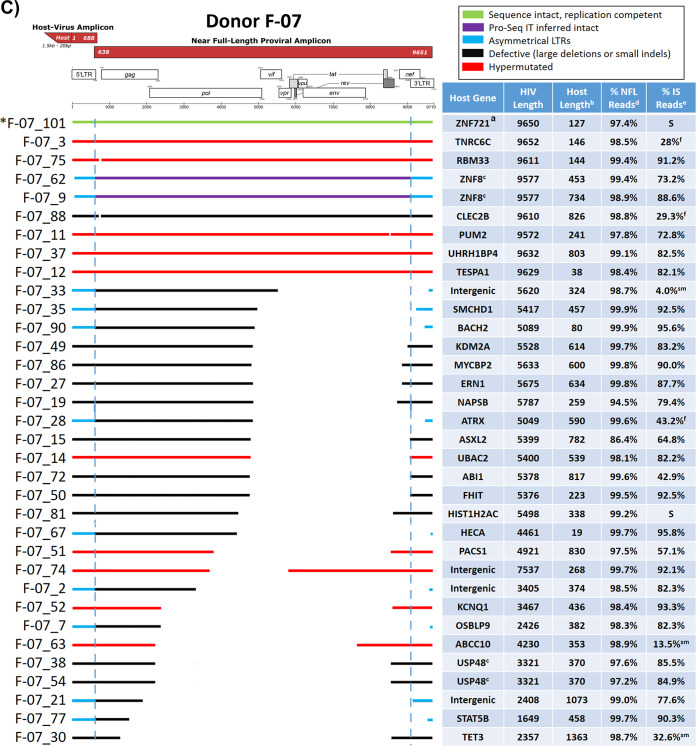
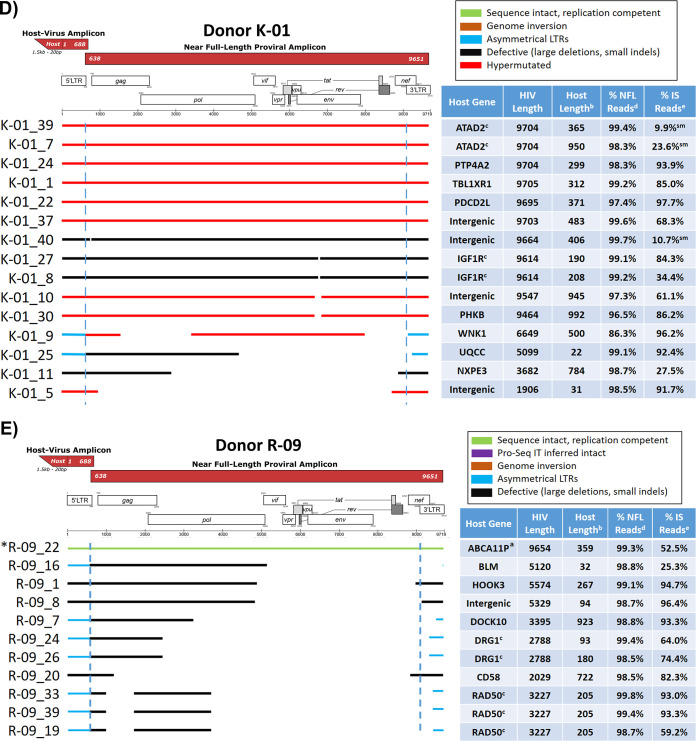
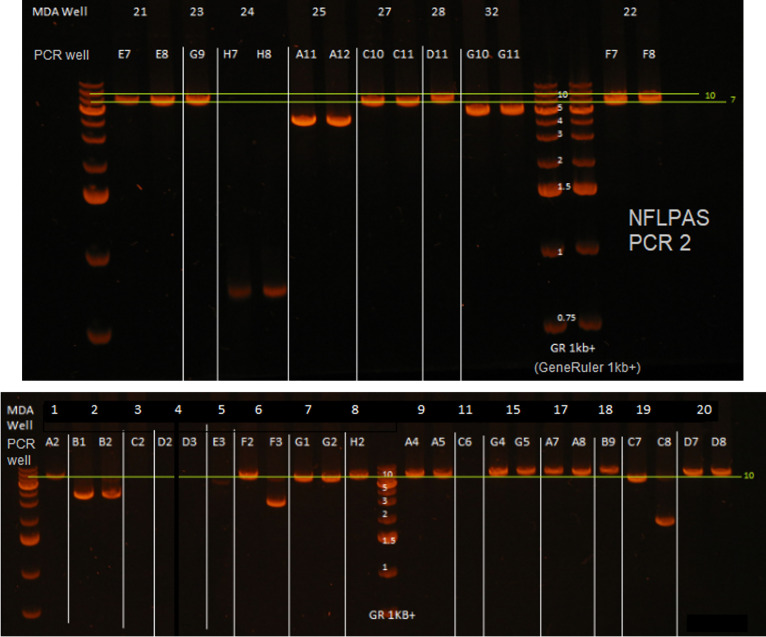
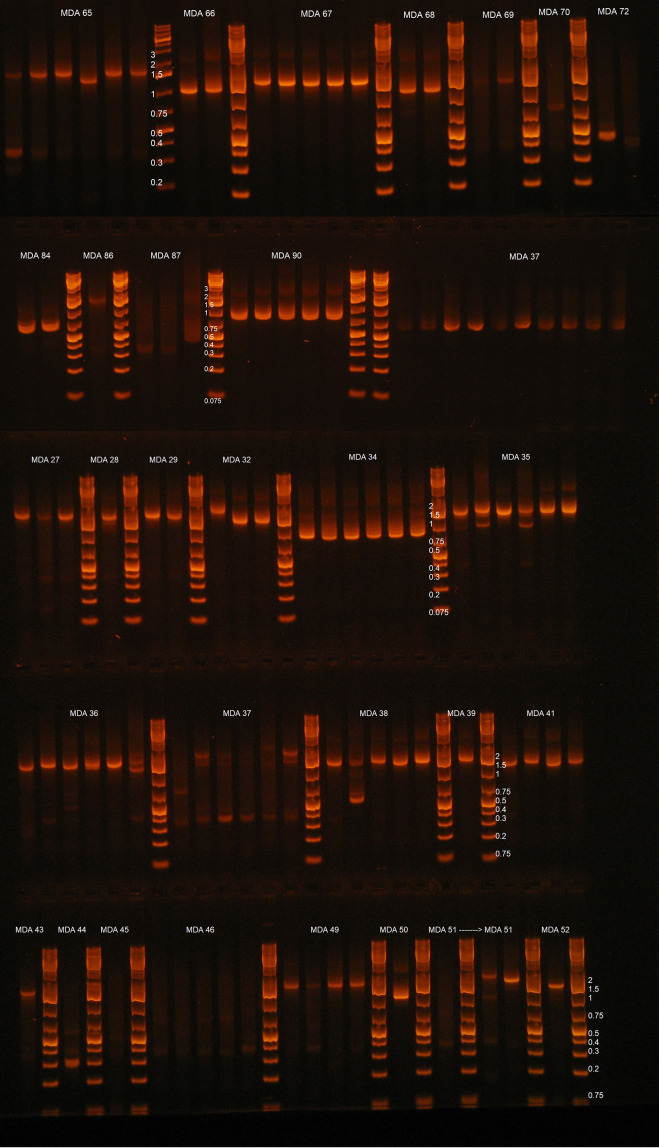
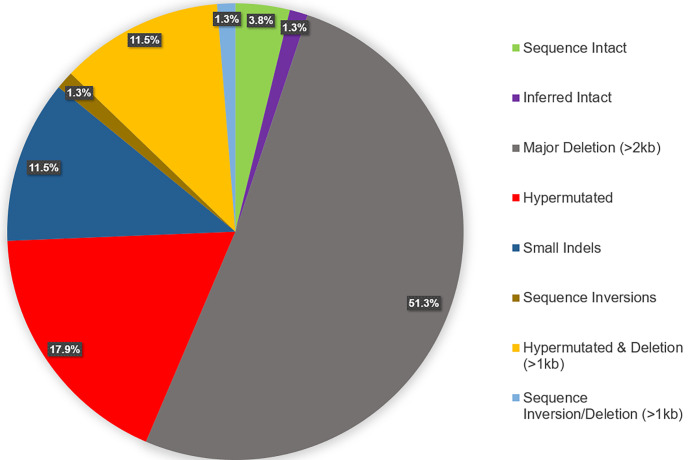
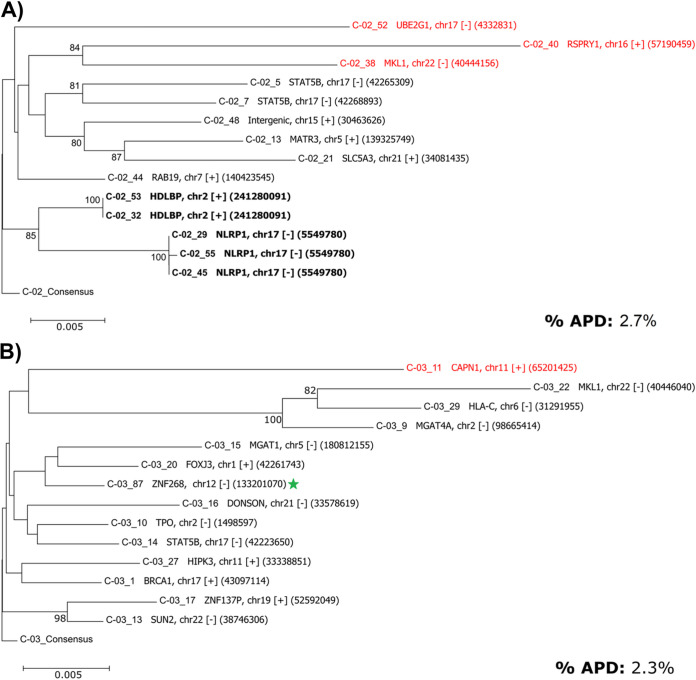
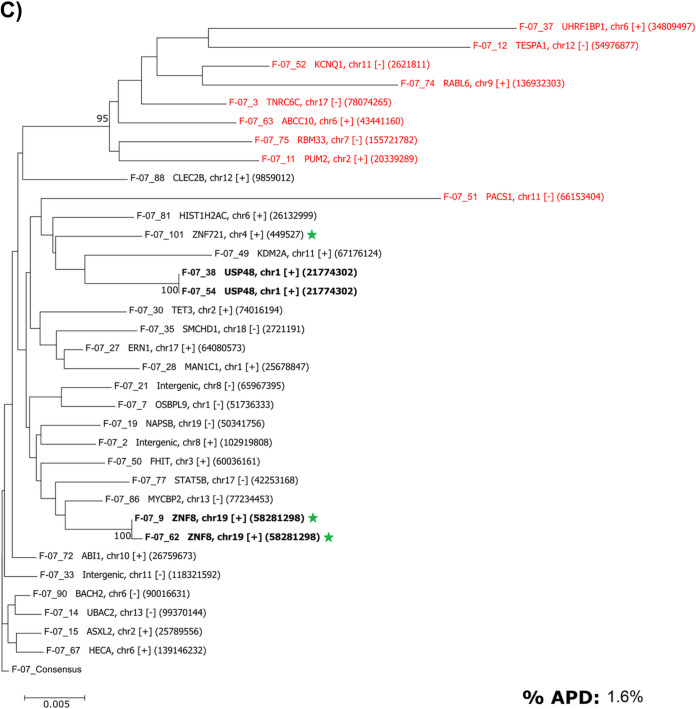
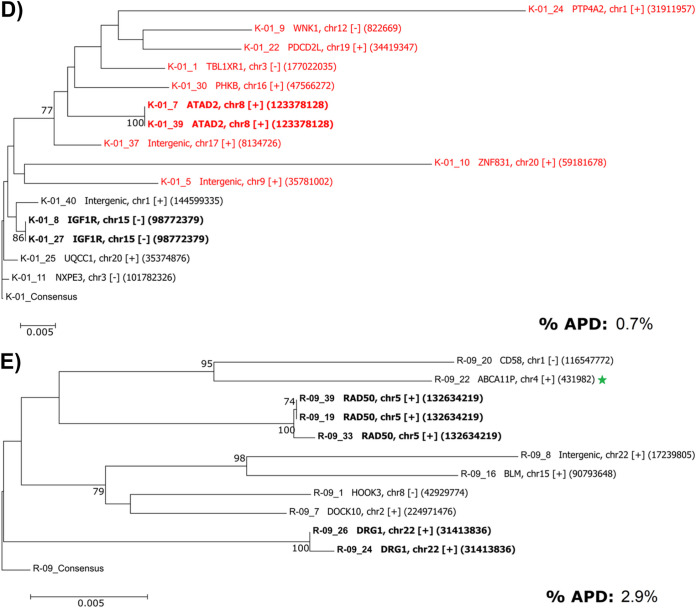
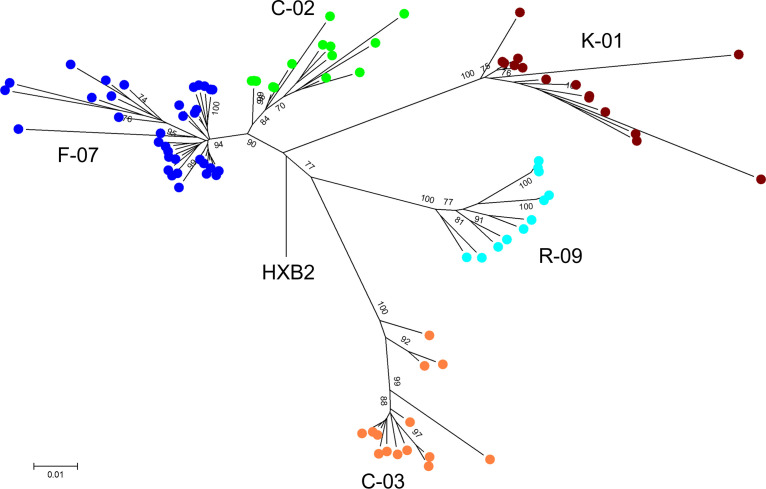
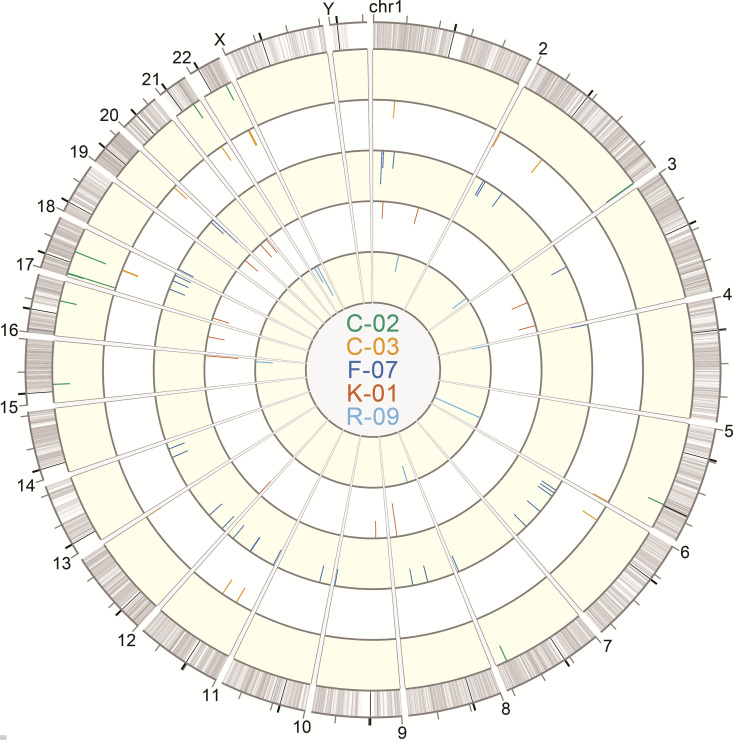
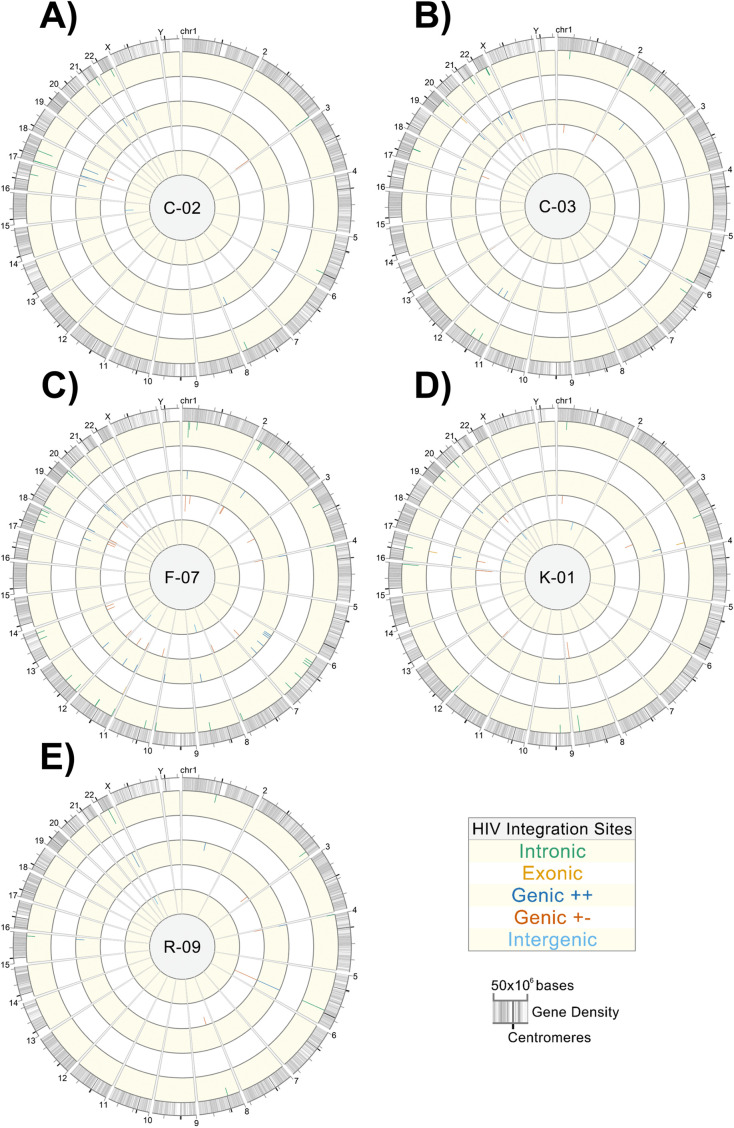
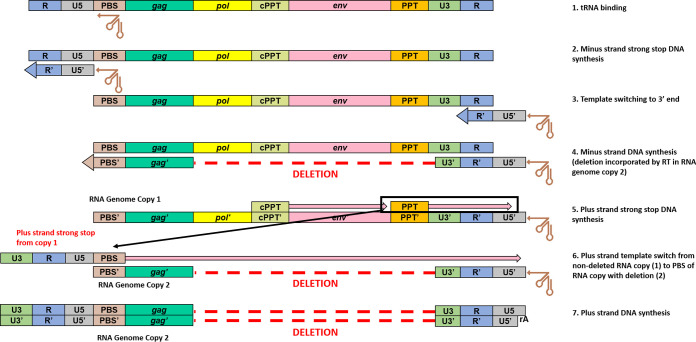
Effective strategies to eliminate human immunodeficiency virus type 1 (HIV-1) reservoirs are likely to require more thorough characterizations of proviruses that persist on antiretroviral therapy (ART). The rarity of infected CD4+ T-cells and related technical challenges have limited the characterization of integrated proviruses. Current approaches using next-generation sequencing can be inefficient and limited sequencing depth can make it difficult to link proviral sequences to their respective integration sites. Here, we report on an efficient method by which HIV-1 proviruses and their sites of integration are amplified and sequenced. Across five HIV-1-positive individuals on clinically effective ART, a median of 41.2% (n = 88 of 209) of amplifications yielded near-full-length proviruses and their 5'-host-virus junctions containing a median of 430 bp (range, 18 to 1,363 bp) of flanking host sequence. Unexpectedly, 29.5% (n = 26 of 88) of the sequenced proviruses had structural asymmetries between the 5' and 3' long terminal repeats (LTRs), commonly in the form of major 3' deletions. Sequence-intact proviruses were detected in 3 of 5 donors, and infected CD4+ T-cell clones were detected in 4 of 5 donors. The accuracy of the method was validated by amplifying and sequencing full-length proviruses and flanking host sequences directly from peripheral blood mononuclear cell DNA. The individual proviral sequencing assay (IPSA) described here can provide an accurate, in-depth, and longitudinal characterization of HIV-1 proviruses that persist on ART, which is important for targeting proviruses for elimination and assessing the impact of interventions designed to eradicate HIV-1. IMPORTANCE The integration of human immunodeficiency virus type 1 (HIV-1) into chromosomal DNA establishes the long-term persistence of HIV-1 as proviruses despite effective antiretroviral therapy (ART). Characterizing proviruses is difficult because of their rarity in individuals on long-term suppressive ART, their highly polymorphic sequences and genetic structures, and the need for efficient amplification and sequencing of the provirus and its integration site. Here, we describe a novel, integrated, two-step method (individual proviral sequencing assay [IPSA]) that amplifies the host-virus junction and the full-length provirus except for the last 69 bp of the 3' long terminal repeat (LTR). Using this method, we identified the integration sites of proviruses, including those that are sequence intact and replication competent or defective. Importantly, this new method identified previously unreported asymmetries between LTRs that have implications for how proviruses are detected and quantified. The IPSA method reported is unaffected by LTR asymmetries, permitting a more accurate and comprehensive characterization of the proviral landscape.
Keywords: HIV-1; LTR; integration sites; proviral structures; sequencing; single genome.
Conflict of interest statement
The authors declare a conflict of interest. J.W.M. reports research grant support for this project from the National Cancer Institute (NCI)/Leidos, National Institutes of Health (NIH), under Contract No. 75N91019D00024, Task Order No. 75N91020F00003; the AIDS Clinical Trials Group Network (ACTG) to the University of Pittsburgh Virology Specialty Laboratory funded by (NIH)/National Institute of Allergy and Infectious Diseases (NIAID) under award UM1 AI106701; and, receives grant support from the Bill & Melinda Gates Foundation award OPP1115715; NIH/NIAID to the I4C (contract numbers UM1 AI126603 and UM1 AI164556); and, REACH (contract number UM1 AI164565) Martin Delaney Collaboratories. M.F.K. and J.W.R. receive grant support from the Office of AIDS Research and from the Intramural Research Programs of the NCI, NIH. J.M.C. receives grant support from The American Cancer Society and NCI through Leidos Subcontract No. l3XS110. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Figures

References
-
- WHO. 2020. HIV data and statistics. WHO, Geneva, Switzerland.
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
