

A reference tissue atlas for the human kidney
- PMID: 35675394
- PMCID: PMC9176741
- DOI: 10.1126/sciadv.abn4965
A reference tissue atlas for the human kidney
Abstract
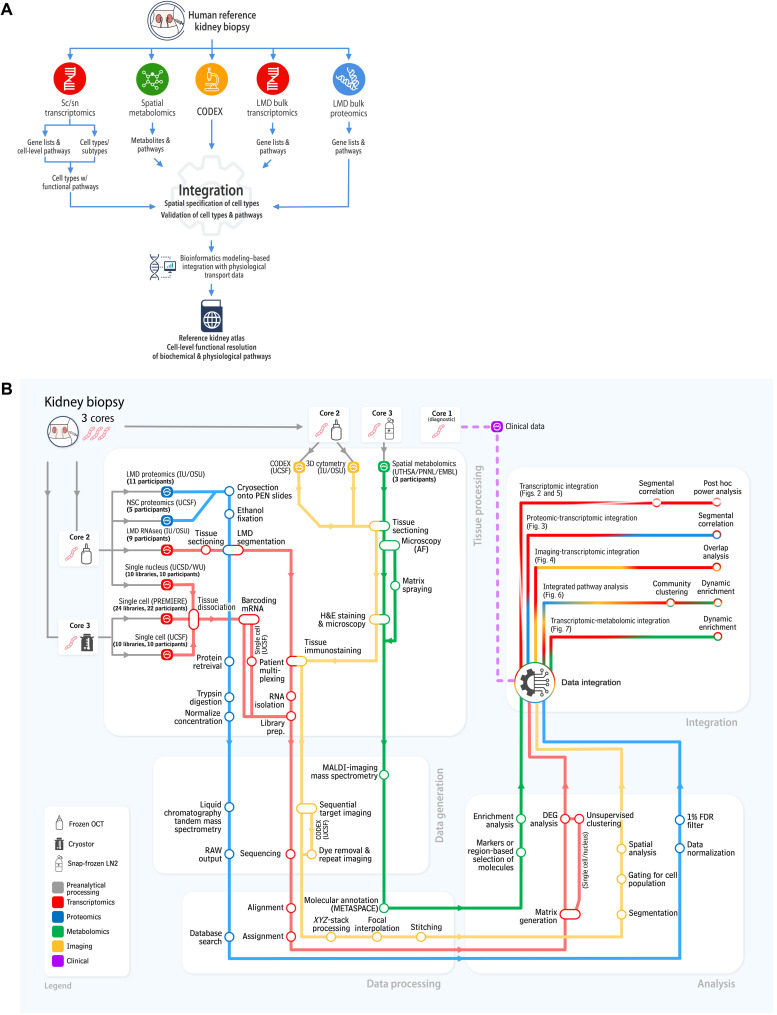
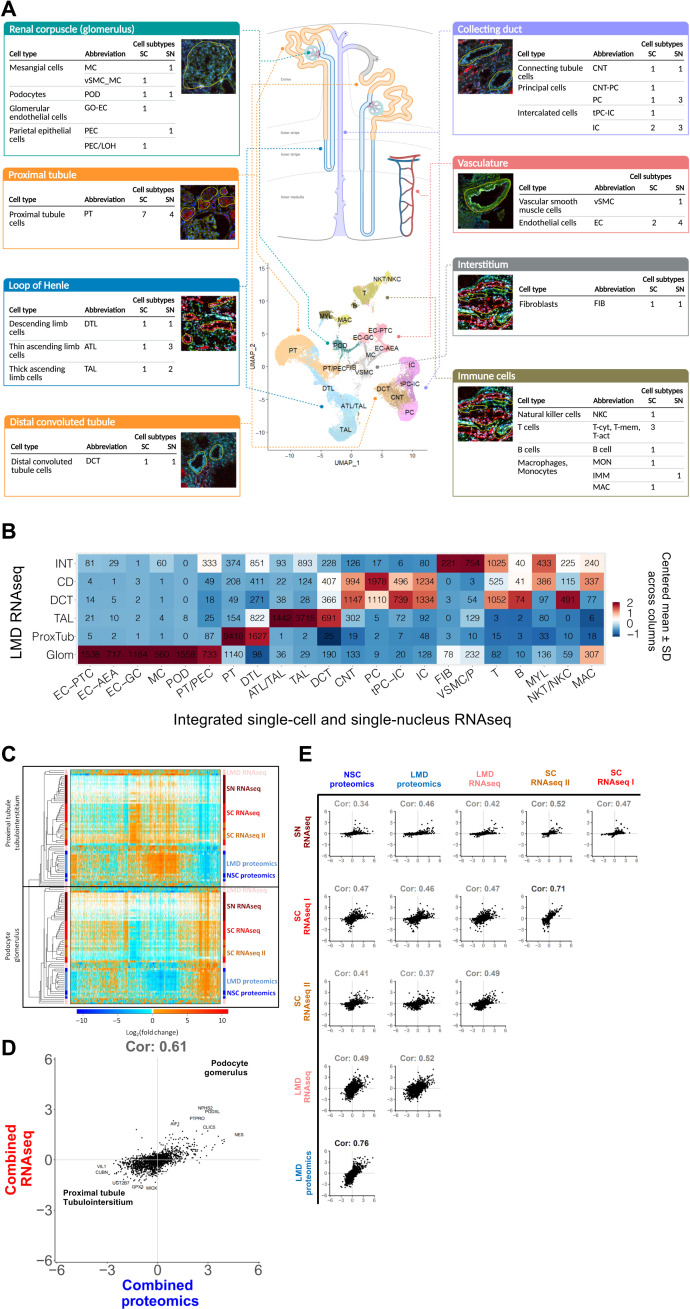
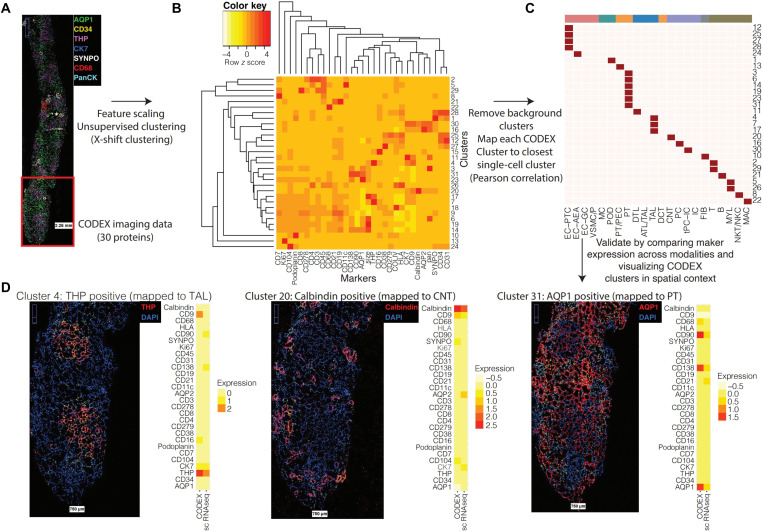
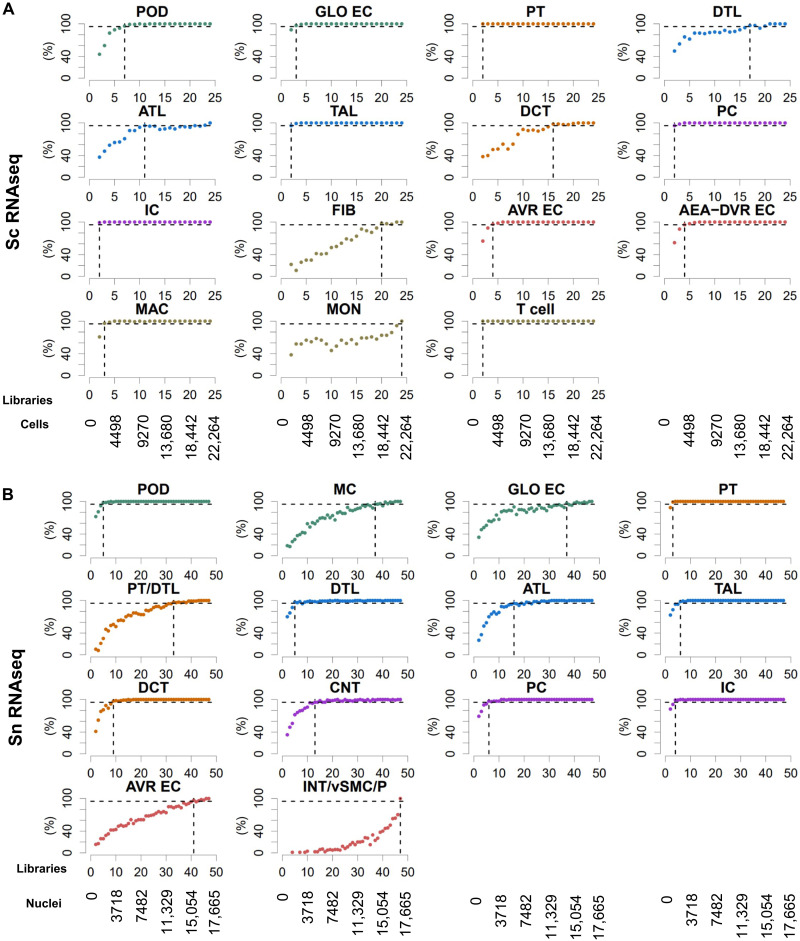
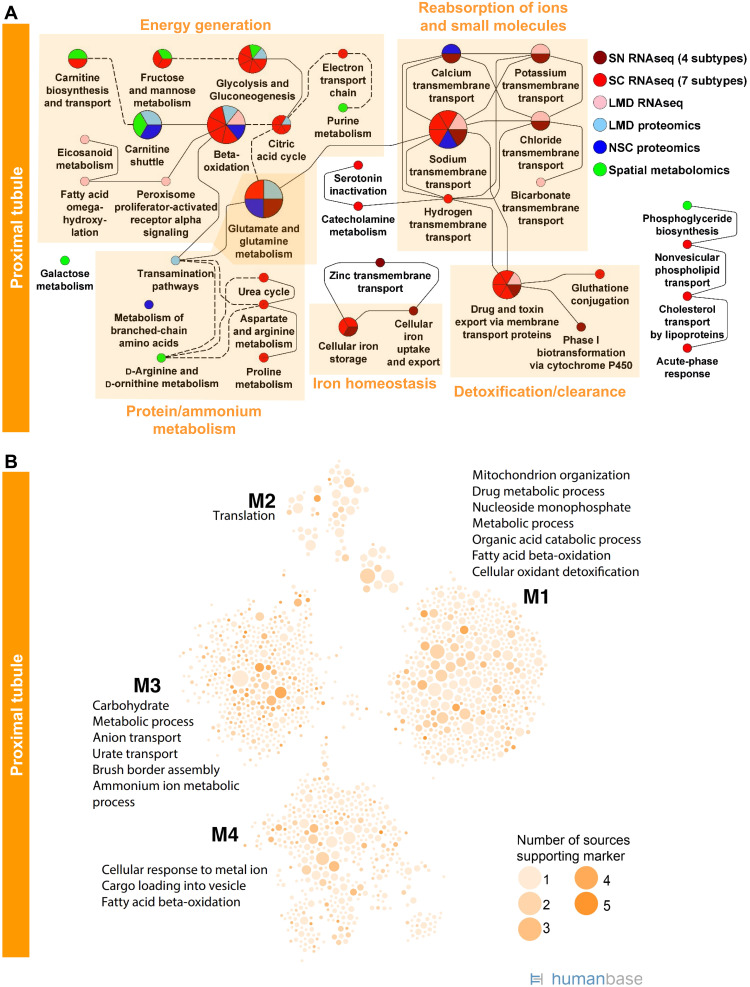
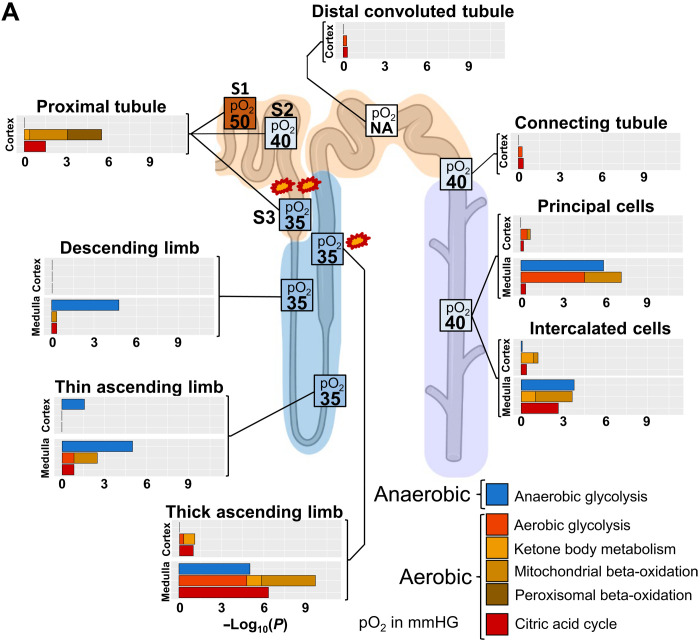
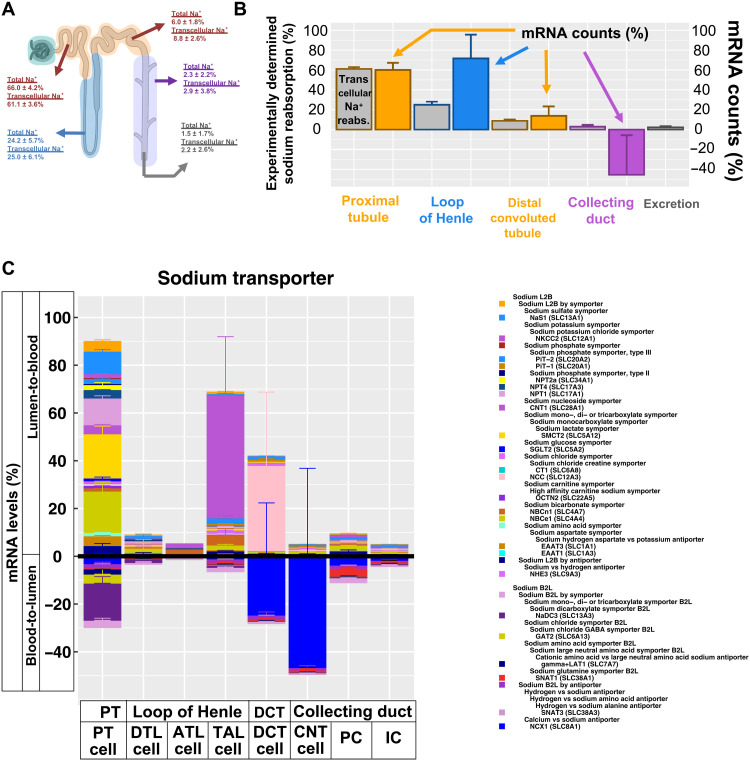
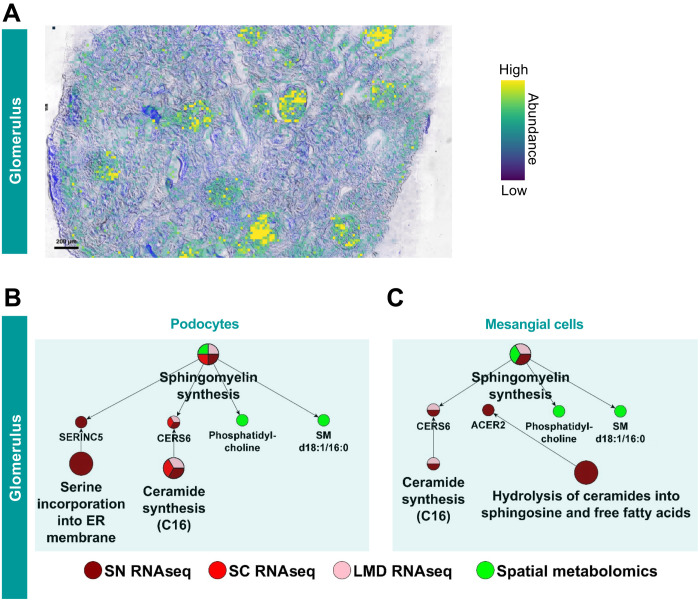
Kidney Precision Medicine Project (KPMP) is building a spatially specified human kidney tissue atlas in health and disease with single-cell resolution. Here, we describe the construction of an integrated reference map of cells, pathways, and genes using unaffected regions of nephrectomy tissues and undiseased human biopsies from 56 adult subjects. We use single-cell/nucleus transcriptomics, subsegmental laser microdissection transcriptomics and proteomics, near-single-cell proteomics, 3D and CODEX imaging, and spatial metabolomics to hierarchically identify genes, pathways, and cells. Integrated data from these different technologies coherently identify cell types/subtypes within different nephron segments and the interstitium. These profiles describe cell-level functional organization of the kidney following its physiological functions and link cell subtypes to genes, proteins, metabolites, and pathways. They further show that messenger RNA levels along the nephron are congruent with the subsegmental physiological activity. This reference atlas provides a framework for the classification of kidney disease when multiple molecular mechanisms underlie convergent clinical phenotypes.
Figures
References
-
- Conway B. R., O’Sullivan E. D., Cairns C., O’Sullivan J., Simpson D. J., Salzano A., Connor K., Ding P., Humphries D., Stewart K., Teenan O., Pius R., Henderson N. C., Bénézech C., Ramachandran P., Ferenbach D., Hughes J., Chandra T., Denby L., Kidney single-cell atlas reveals myeloid heterogeneity in progression and regression of kidney disease. J. Am. Soc. Nephrol. 31, 2833–2854 (2020). - PMC - PubMed
-
- He B., Chen P., Zambrano S., Dabaghie D., Hu Y., Möller-Hackbarth K., Unnersjö-Jess D., Korkut G. G., Charrin E., Jeansson M., Bintanel-Morcillo M., Witasp A., Wennberg L., Wernerson A., Schermer B., Benzing T., Ernfors P., Betsholtz C., Lal M., Sandberg R., Patrakka J., Single-cell RNA sequencing reveals the mesangial identity and species diversity of glomerular cell transcriptomes. Nat. Commun. 12, 2141 (2021). - PMC - PubMed
-
- Huang L., Liao J., He J., Pan S., Zhang H., Yang X., Cheng J., Chen Y., Mo Z., Single-cell profiling reveals sex diversity in human renal PTs. Gene 752, 144790 (2020). - PubMed
Publication types
MeSH terms
Grants and funding
- UH3 DK114907/DK/NIDDK NIH HHS/United States
- UG3 DK114937/DK/NIDDK NIH HHS/United States
- U2C DK114886/DK/NIDDK NIH HHS/United States
- UH3 DK114920/DK/NIDDK NIH HHS/United States
- U01 DK114933/DK/NIDDK NIH HHS/United States
- U54 HL145608/HL/NHLBI NIH HHS/United States
- P30 DK079312/DK/NIDDK NIH HHS/United States
- UH3 DK114937/DK/NIDDK NIH HHS/United States
- U01 DK114907/DK/NIDDK NIH HHS/United States
- UG3 DK114907/DK/NIDDK NIH HHS/United States
- UH3 DK114933/DK/NIDDK NIH HHS/United States
- U24 DK114886/DK/NIDDK NIH HHS/United States
- UH3 DK114923/DK/NIDDK NIH HHS/United States
- K23 DK125529/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
