

Phenotypic and genetic spectrum of ATP6V1A encephalopathy: a disorder of lysosomal homeostasis
- PMID: 35675510
- PMCID: PMC10893886
- DOI: 10.1093/brain/awac145
Phenotypic and genetic spectrum of ATP6V1A encephalopathy: a disorder of lysosomal homeostasis
Abstract
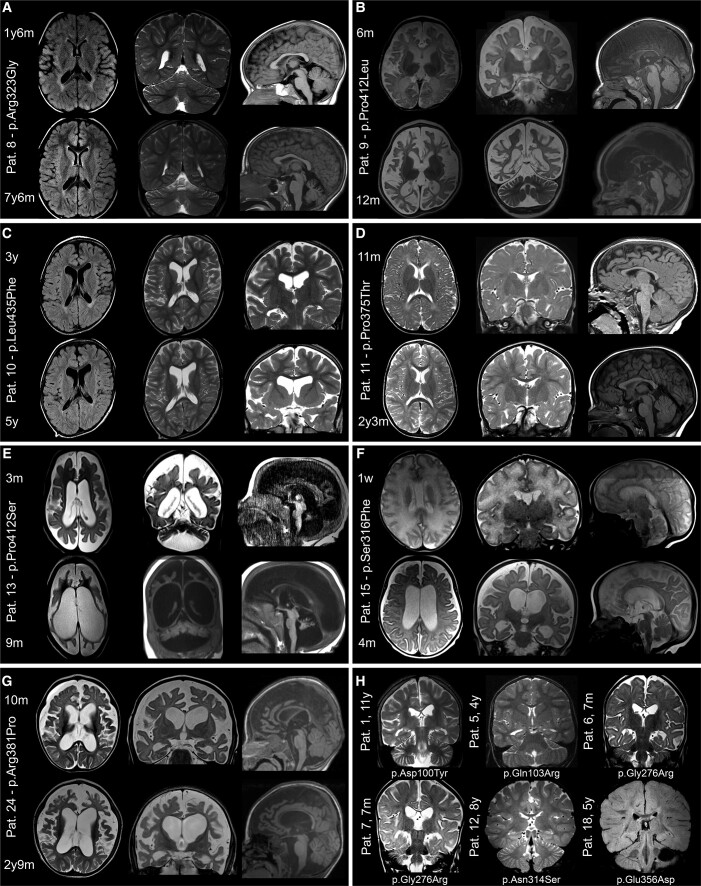
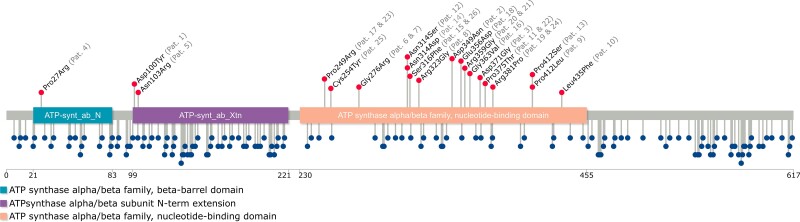
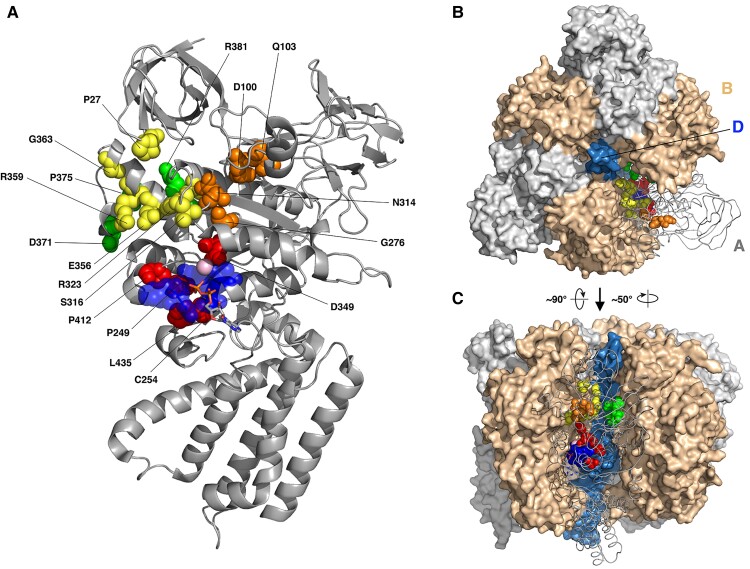
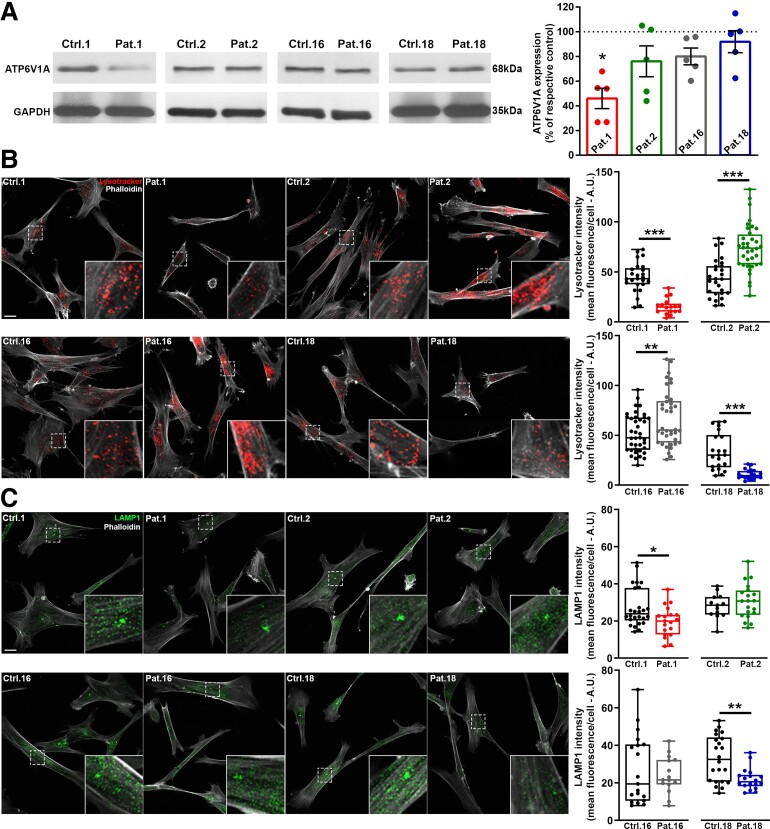
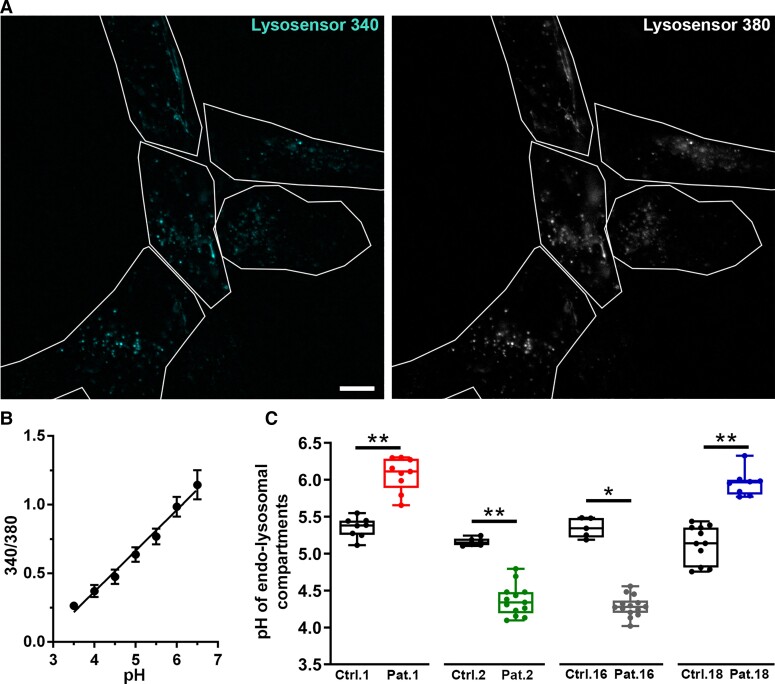
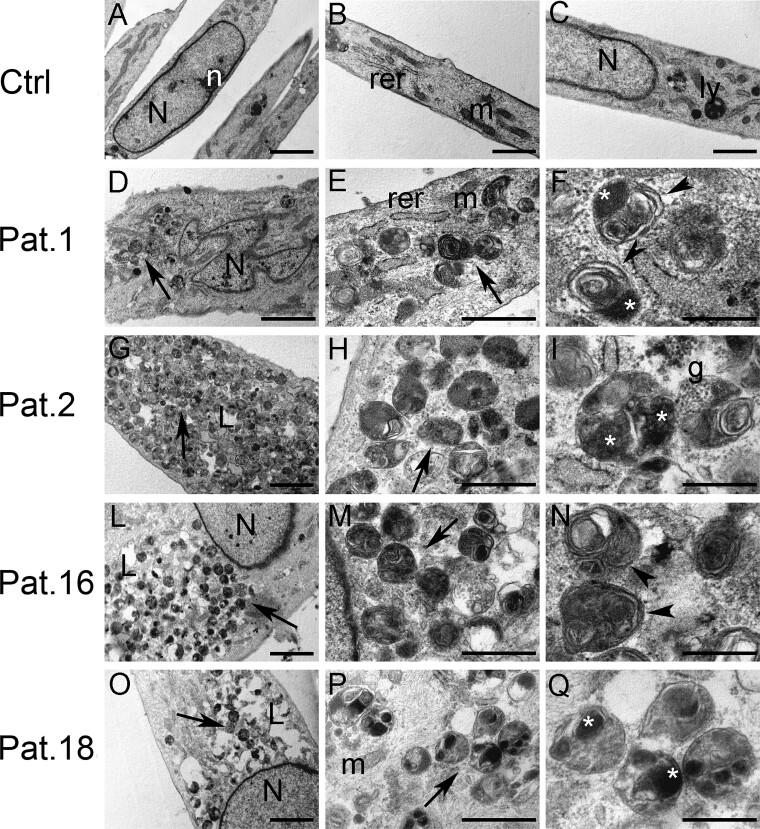
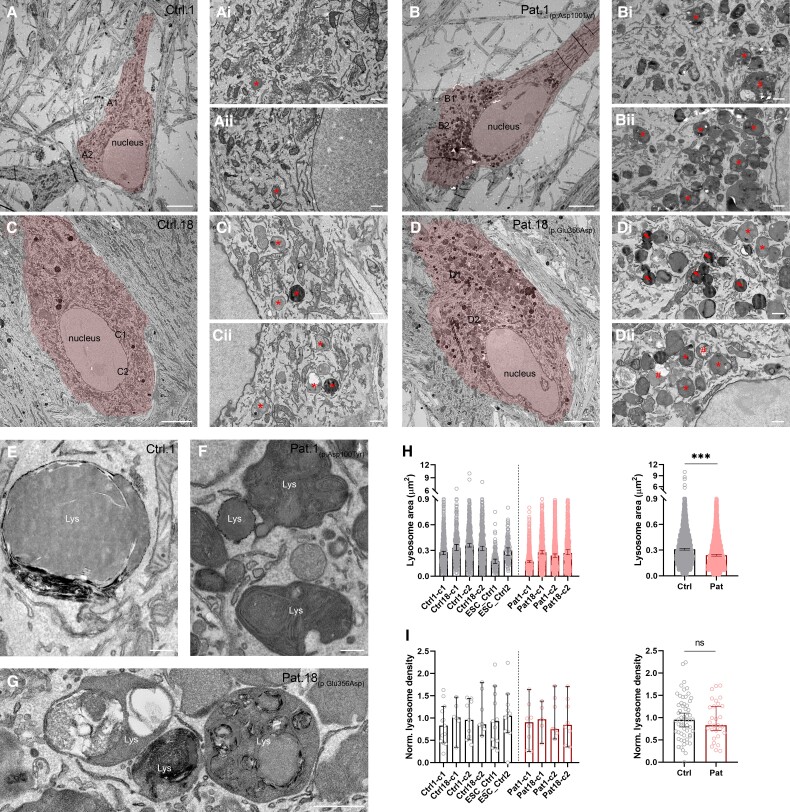
Vacuolar-type H+-ATPase (V-ATPase) is a multimeric complex present in a variety of cellular membranes that acts as an ATP-dependent proton pump and plays a key role in pH homeostasis and intracellular signalling pathways. In humans, 22 autosomal genes encode for a redundant set of subunits allowing the composition of diverse V-ATPase complexes with specific properties and expression. Sixteen subunits have been linked to human disease. Here we describe 26 patients harbouring 20 distinct pathogenic de novo missense ATP6V1A variants, mainly clustering within the ATP synthase α/β family-nucleotide-binding domain. At a mean age of 7 years (extremes: 6 weeks, youngest deceased patient to 22 years, oldest patient) clinical pictures included early lethal encephalopathies with rapidly progressive massive brain atrophy, severe developmental epileptic encephalopathies and static intellectual disability with epilepsy. The first clinical manifestation was early hypotonia, in 70%; 81% developed epilepsy, manifested as developmental epileptic encephalopathies in 58% of the cohort and with infantile spasms in 62%; 63% of developmental epileptic encephalopathies failed to achieve any developmental, communicative or motor skills. Less severe outcomes were observed in 23% of patients who, at a mean age of 10 years and 6 months, exhibited moderate intellectual disability, with independent walking and variable epilepsy. None of the patients developed communicative language. Microcephaly (38%) and amelogenesis imperfecta/enamel dysplasia (42%) were additional clinical features. Brain MRI demonstrated hypomyelination and generalized atrophy in 68%. Atrophy was progressive in all eight individuals undergoing repeated MRIs. Fibroblasts of two patients with developmental epileptic encephalopathies showed decreased LAMP1 expression, Lysotracker staining and increased organelle pH, consistent with lysosomal impairment and loss of V-ATPase function. Fibroblasts of two patients with milder disease, exhibited a different phenotype with increased Lysotracker staining, decreased organelle pH and no significant modification in LAMP1 expression. Quantification of substrates for lysosomal enzymes in cellular extracts from four patients revealed discrete accumulation. Transmission electron microscopy of fibroblasts of four patients with variable severity and of induced pluripotent stem cell-derived neurons from two patients with developmental epileptic encephalopathies showed electron-dense inclusions, lipid droplets, osmiophilic material and lamellated membrane structures resembling phospholipids. Quantitative assessment in induced pluripotent stem cell-derived neurons identified significantly smaller lysosomes. ATP6V1A-related encephalopathy represents a new paradigm among lysosomal disorders. It results from a dysfunctional endo-lysosomal membrane protein causing altered pH homeostasis. Its pathophysiology implies intracellular accumulation of substrates whose composition remains unclear, and a combination of developmental brain abnormalities and neurodegenerative changes established during prenatal and early postanal development, whose severity is variably determined by specific pathogenic variants.
Keywords: ATP6V1A; developmental delay; epileptic encephalopathy; lysosomal disorder; progressive brain atrophy.
© The Author(s) 2022. Published by Oxford University Press on behalf of the Guarantors of Brain. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
Conflict of interest statement
The Department of Molecular and Human Genetics at Baylor College of Medicine receives revenue from clinical genetic testing conducted at Baylor Genetics Laboratories (J.A.R., C.A.B.). Y.S., I.M.W. and F.Z. are employees of GeneDx, Inc. Z.P. is a past employee of Ambry Genetics. All other authors declare they have no competing interests.
Figures
Comment in
-
Rare human ATP6V1A variants provide unique insights into V-ATPase functions.Brain. 2022 Aug 27;145(8):2626-2628. doi: 10.1093/brain/awac255. Brain. 2022. PMID: 35857819 No abstract available.
References
-
- Forgac M. Vacuolar ATPases: Rotary proton pumps in physiology and pathophysiology. Nat Rev Mol Cell Biol. 2007;8:917–929. - PubMed
-
- Bodzęta A, Kahms M, Klingauf J. The presynaptic v-ATPase reversibly disassembles and thereby modulates exocytosis but is not part of the fusion machinery. Cell Rep. 2017;20:1348–1359. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
