

Mature neutrophils and a NF-κB-to-IFN transition determine the unifying disease recovery dynamics in COVID-19
- PMID: 35675822
- PMCID: PMC9110324
- DOI: 10.1016/j.xcrm.2022.100652
Mature neutrophils and a NF-κB-to-IFN transition determine the unifying disease recovery dynamics in COVID-19
Abstract
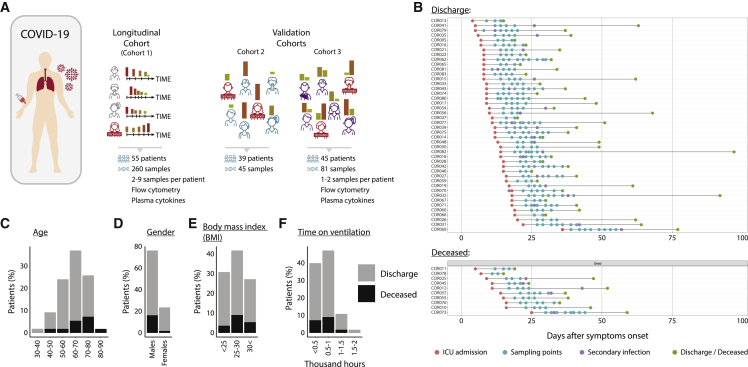
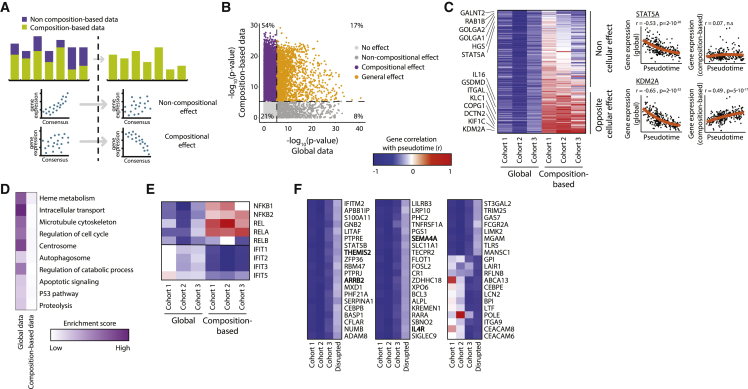
Disease recovery dynamics are often difficult to assess, as patients display heterogeneous recovery courses. To model recovery dynamics, exemplified by severe COVID-19, we apply a computational scheme on longitudinally sampled blood transcriptomes, generating recovery states, which we then link to cellular and molecular mechanisms, presenting a framework for studying the kinetics of recovery compared with non-recovery over time and long-term effects of the disease. Specifically, a decrease in mature neutrophils is the strongest cellular effect during recovery, with direct implications on disease outcome. Furthermore, we present strong indications for global regulatory changes in gene programs, decoupled from cell compositional changes, including an early rise in T cell activation and differentiation, resulting in immune rebalancing between interferon and NF-κB activity and restoration of cell homeostasis. Overall, we present a clinically relevant computational framework for modeling disease recovery, paving the way for future studies of the recovery dynamics in other diseases and tissues.
Keywords: COVID-19; cell deconvolution; disease modeling; disease recovery; gene regulation; immunology; medicine; systems biology; viral infection.
Copyright © 2022 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests F.J.T. reports receiving consulting fees from ImmunAI and ownership interest in Dermagnostix. S.S.S.-O. holds equity and is a consultant of CytoReason.
Figures




References
-
- Silvin A., Chapuis N., Dunsmore G., Goubet A.-G., Dubuisson A., Derosa L., Almire C., Hénon C., Kosmider O., Droin N., et al. Elevated calprotectin and abnormal myeloid cell subsets discriminate severe from mild COVID-19. Cell. 2020;182:1401–1418.e18. doi: 10.1016/j.cell.2020.08.002. - DOI - PMC - PubMed
-
- Aschenbrenner A.C., Mouktaroudi M., Krämer B., Oestreich M., Antonakos N., Nuesch-Germano M., Gkizeli K., Bonaguro L., Reusch N., Baßler K., et al. Disease severity-specific neutrophil signatures in blood transcriptomes stratify COVID-19 patients. Genome. Med. 2021;13:7. doi: 10.1186/s13073-020-00823-5. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
