

The renal lineage factor PAX8 controls oncogenic signalling in kidney cancer
- PMID: 35676472
- PMCID: PMC9242860
- DOI: 10.1038/s41586-022-04809-8
The renal lineage factor PAX8 controls oncogenic signalling in kidney cancer
Abstract
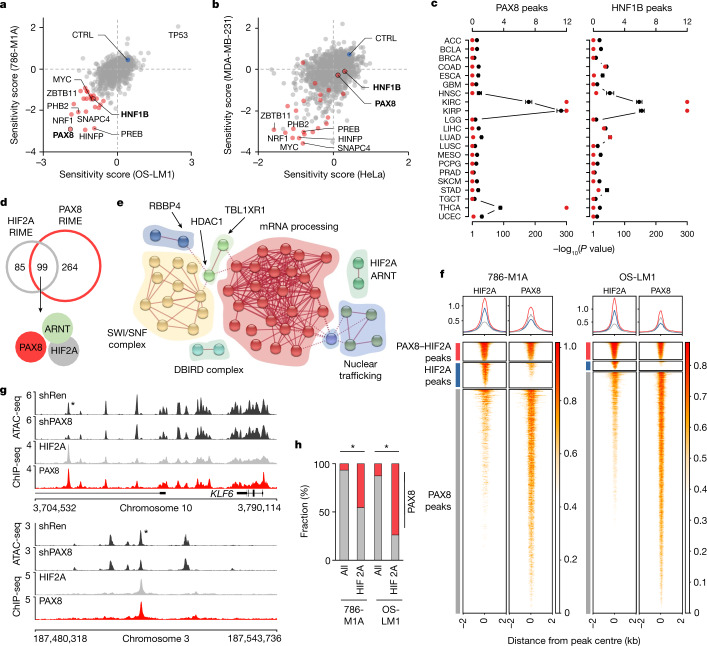
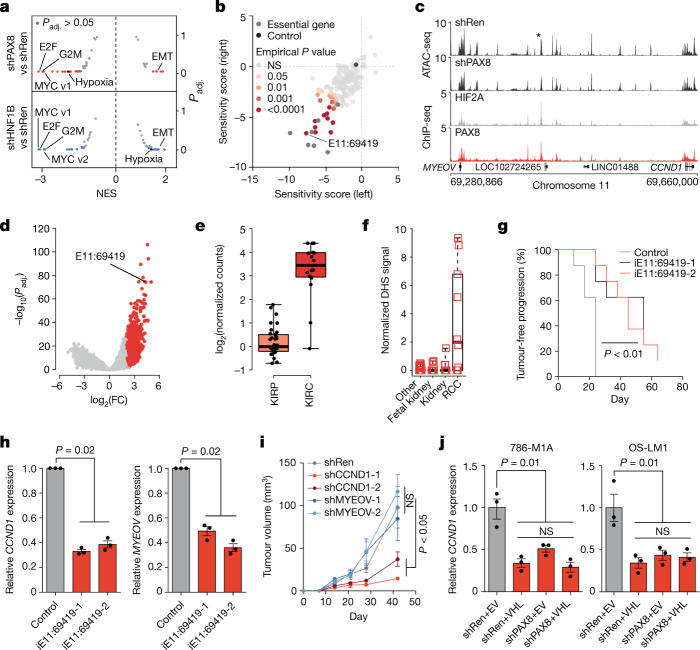
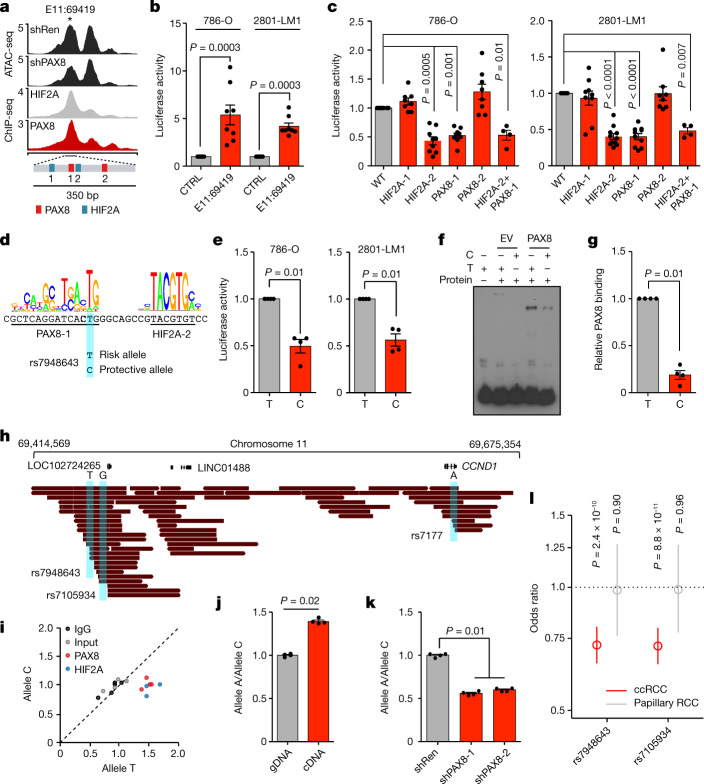
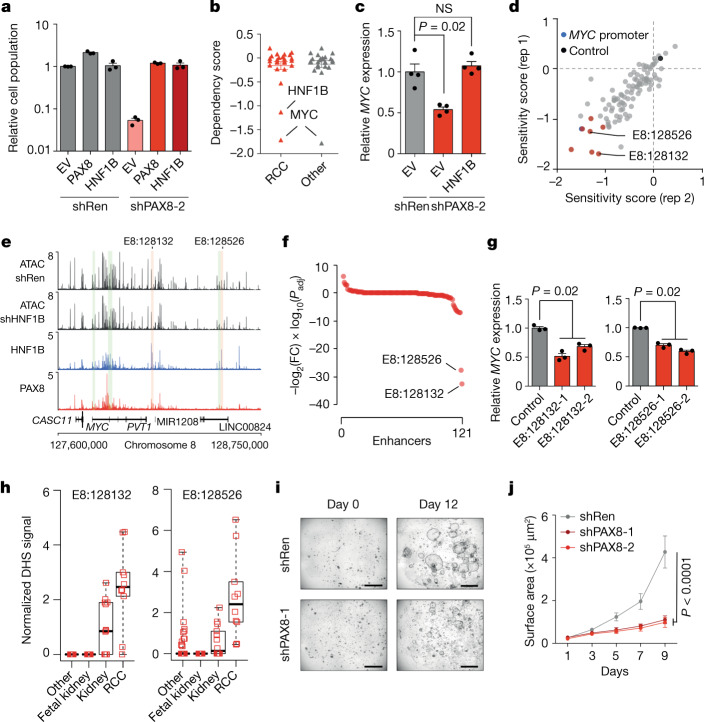
Large-scale human genetic data1-3 have shown that cancer mutations display strong tissue-selectivity, but how this selectivity arises remains unclear. Here, using experimental models, functional genomics and analyses of patient samples, we demonstrate that the lineage transcription factor paired box 8 (PAX8) is required for oncogenic signalling by two common genetic alterations that cause clear cell renal cell carcinoma (ccRCC) in humans: the germline variant rs7948643 at 11q13.3 and somatic inactivation of the von Hippel-Lindau tumour suppressor (VHL)4-6. VHL loss, which is observed in about 90% of ccRCCs, can lead to hypoxia-inducible factor 2α (HIF2A) stabilization6,7. We show that HIF2A is preferentially recruited to PAX8-bound transcriptional enhancers, including a pro-tumorigenic cyclin D1 (CCND1) enhancer that is controlled by PAX8 and HIF2A. The ccRCC-protective allele C at rs7948643 inhibits PAX8 binding at this enhancer and downstream activation of CCND1 expression. Co-option of a PAX8-dependent physiological programme that supports the proliferation of normal renal epithelial cells is also required for MYC expression from the ccRCC metastasis-associated amplicons at 8q21.3-q24.3 (ref. 8). These results demonstrate that transcriptional lineage factors are essential for oncogenic signalling and that they mediate tissue-specific cancer risk associated with somatic and inherited genetic variants.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures











Comment in
-
Mutation and tissue lineage lead to organ-specific cancer.Nature. 2022 Jun;606(7916):871-872. doi: 10.1038/d41586-022-01535-z. Nature. 2022. PMID: 35676353 Free PMC article.
-
Oncogenic Signaling in Kidney Cancer Requires Renal Lineage Factor PAX8.Cancer Discov. 2022 Aug 5;12(8):1834. doi: 10.1158/2159-8290.CD-RW2022-113. Cancer Discov. 2022. PMID: 35713353
-
Control of oncogenic signalling in ccRCC.Nat Rev Urol. 2022 Aug;19(8):453. doi: 10.1038/s41585-022-00627-9. Nat Rev Urol. 2022. PMID: 35831546 No abstract available.
References
-
- Fletcher O, Houlston RS. Architecture of inherited susceptibility to common cancer. Nat. Rev. Cancer. 2010;10:353–361. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
