

Graph pangenome captures missing heritability and empowers tomato breeding
- PMID: 35676474
- PMCID: PMC9200638
- DOI: 10.1038/s41586-022-04808-9
Graph pangenome captures missing heritability and empowers tomato breeding
Abstract
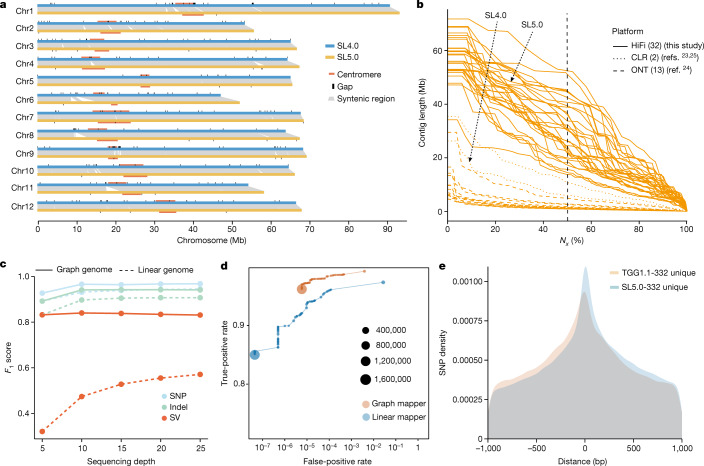
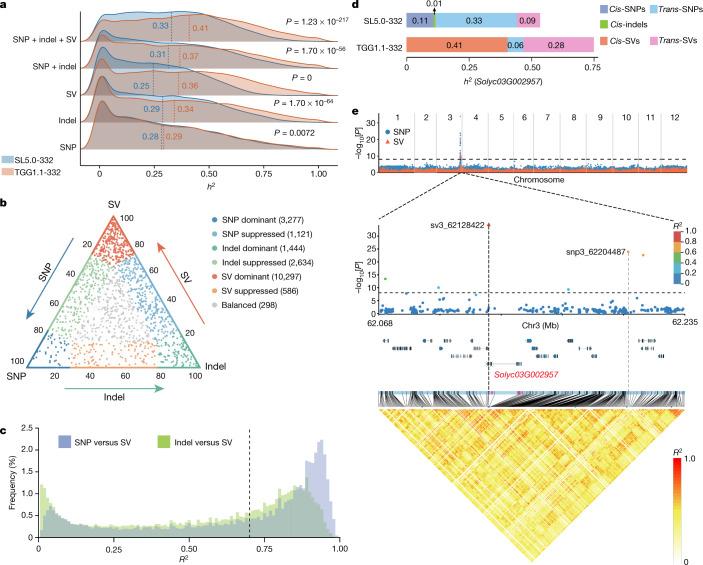
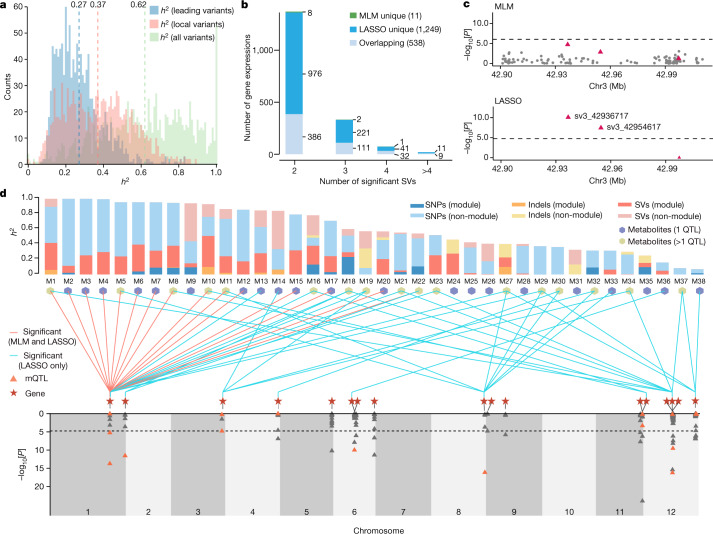
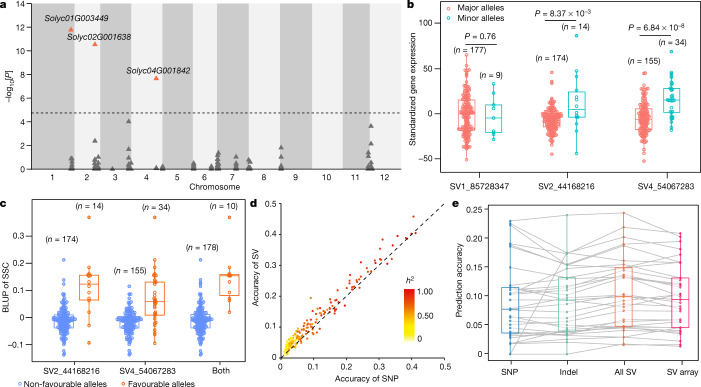
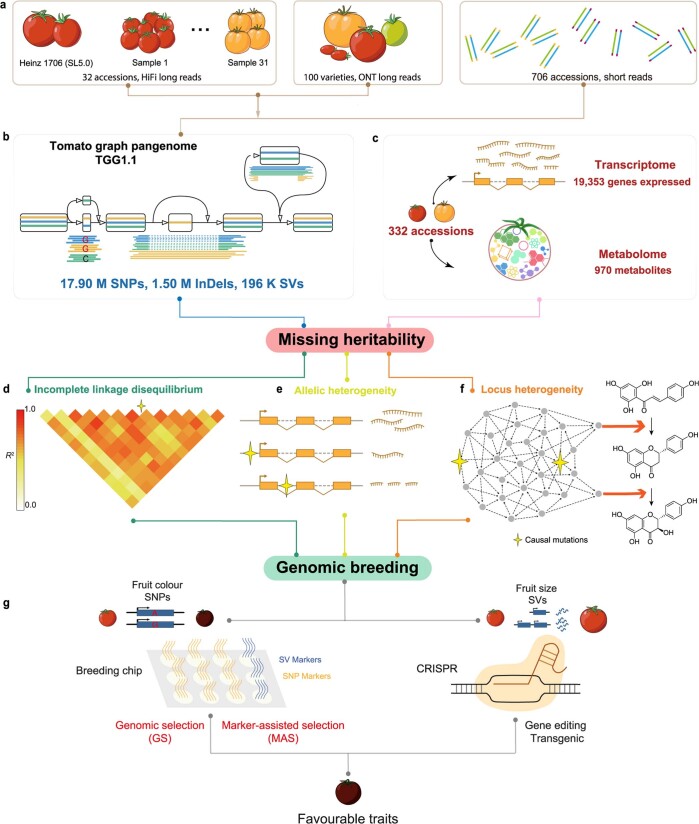
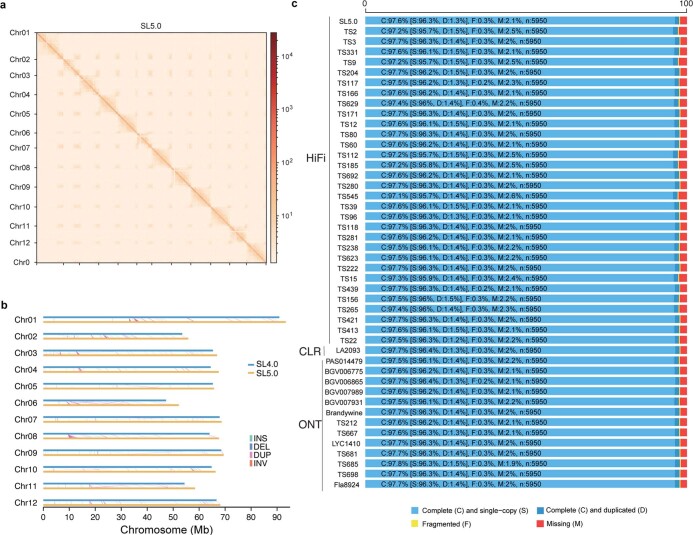
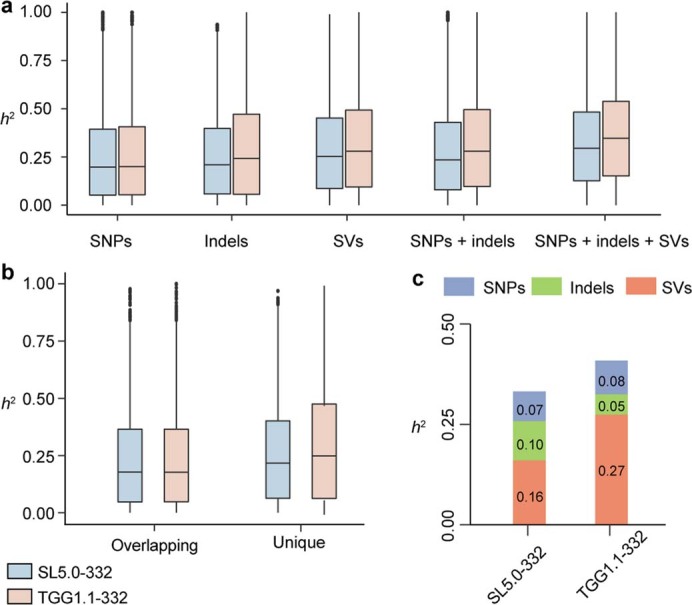
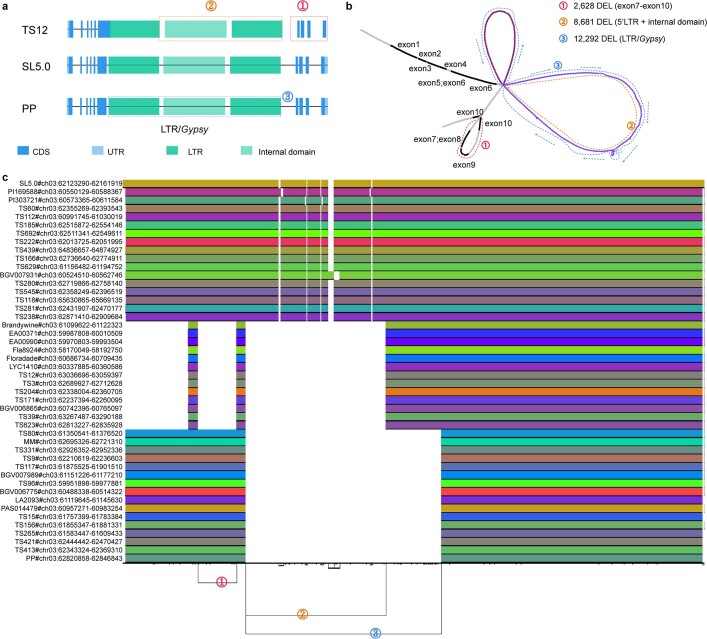
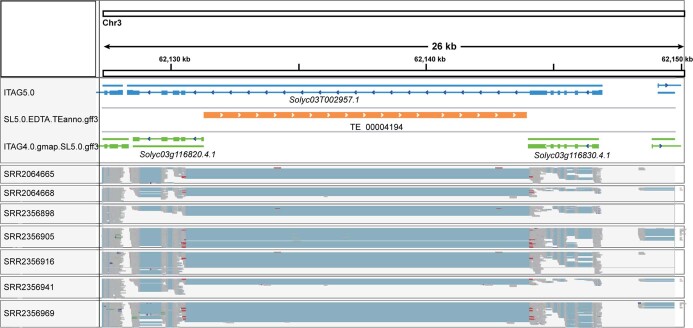
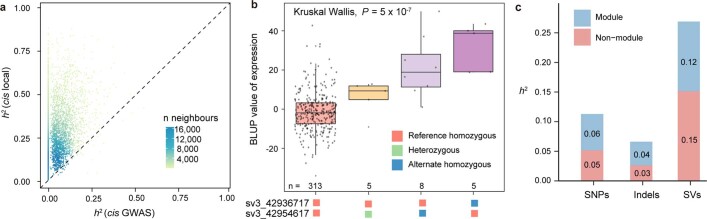
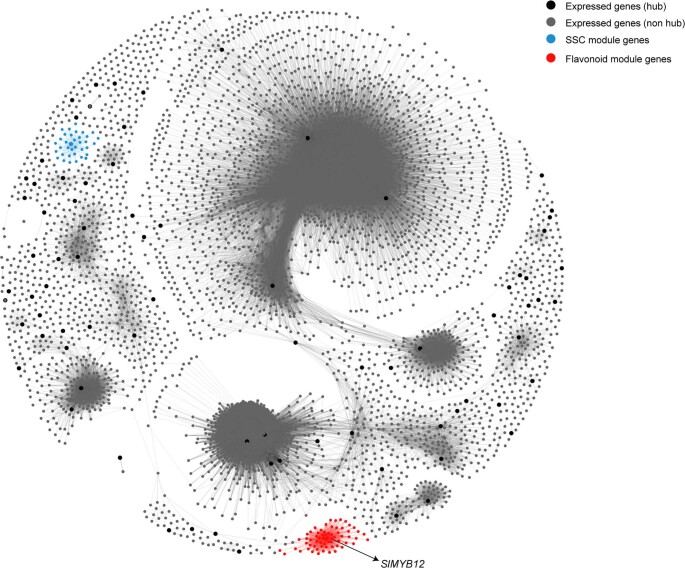
Missing heritability in genome-wide association studies defines a major problem in genetic analyses of complex biological traits1,2. The solution to this problem is to identify all causal genetic variants and to measure their individual contributions3,4. Here we report a graph pangenome of tomato constructed by precisely cataloguing more than 19 million variants from 838 genomes, including 32 new reference-level genome assemblies. This graph pangenome was used for genome-wide association study analyses and heritability estimation of 20,323 gene-expression and metabolite traits. The average estimated trait heritability is 0.41 compared with 0.33 when using the single linear reference genome. This 24% increase in estimated heritability is largely due to resolving incomplete linkage disequilibrium through the inclusion of additional causal structural variants identified using the graph pangenome. Moreover, by resolving allelic and locus heterogeneity, structural variants improve the power to identify genetic factors underlying agronomically important traits leading to, for example, the identification of two new genes potentially contributing to soluble solid content. The newly identified structural variants will facilitate genetic improvement of tomato through both marker-assisted selection and genomic selection. Our study advances the understanding of the heritability of complex traits and demonstrates the power of the graph pangenome in crop breeding.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

Comment in
-
Graph pangenomes find missing heritability.Nat Genet. 2022 Jul;54(7):919-920. doi: 10.1038/s41588-022-01099-8. Nat Genet. 2022. PMID: 35739387 No abstract available.
-
Cutting the Gordian knot of plant genetics: retrieval of missing heritability in tomato.Sci China Life Sci. 2022 Dec;65(12):2564-2566. doi: 10.1007/s11427-022-2160-6. Epub 2022 Jul 15. Sci China Life Sci. 2022. PMID: 35851941 No abstract available.
References
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
