

Cohesin-mediated loop anchors confine the locations of human replication origins
- PMID: 35676475
- PMCID: PMC9217744
- DOI: 10.1038/s41586-022-04803-0
Cohesin-mediated loop anchors confine the locations of human replication origins
Abstract
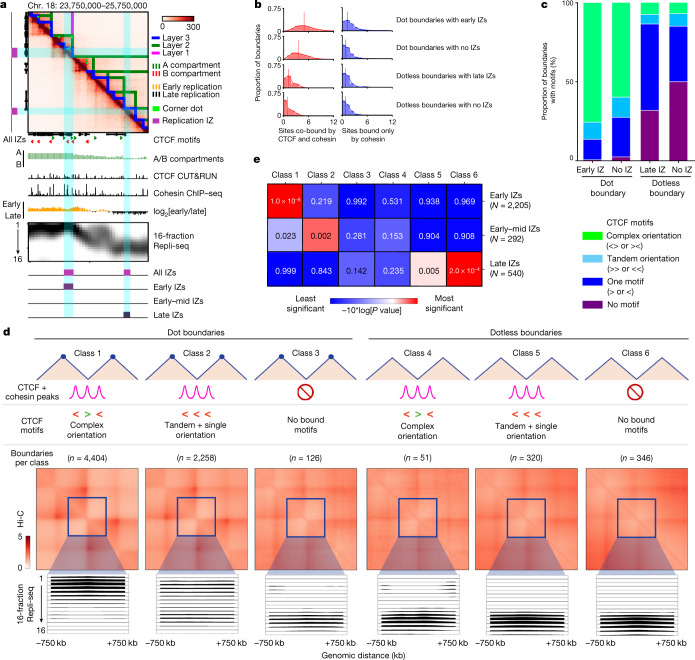
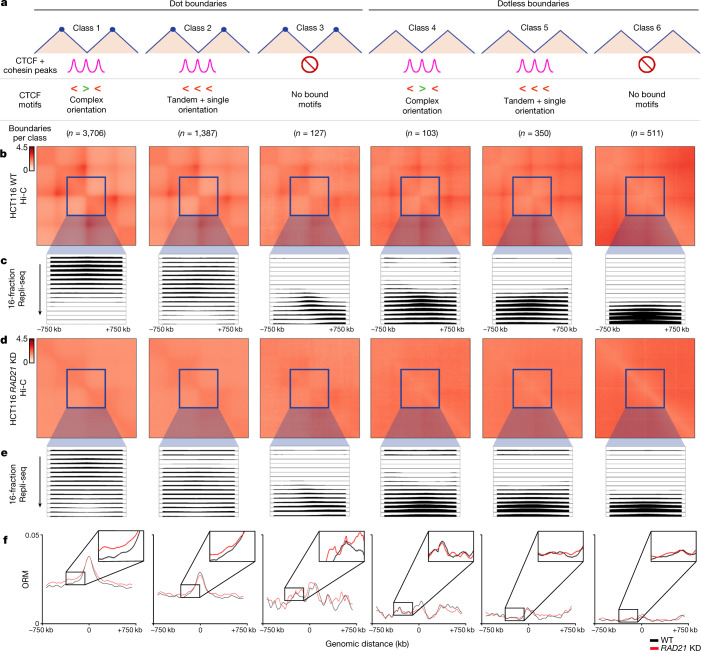
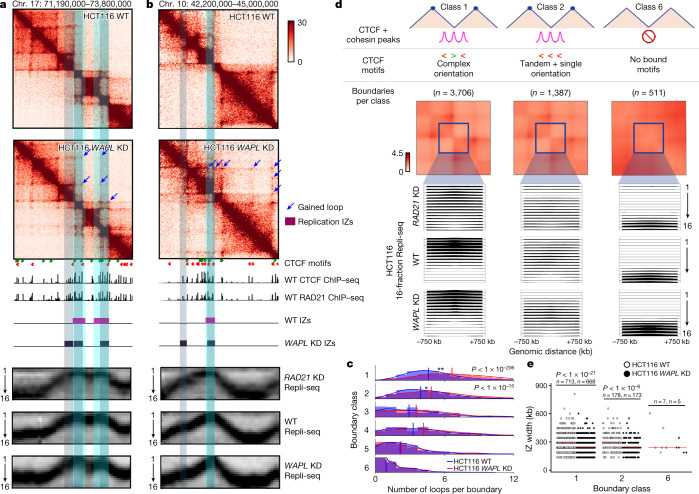
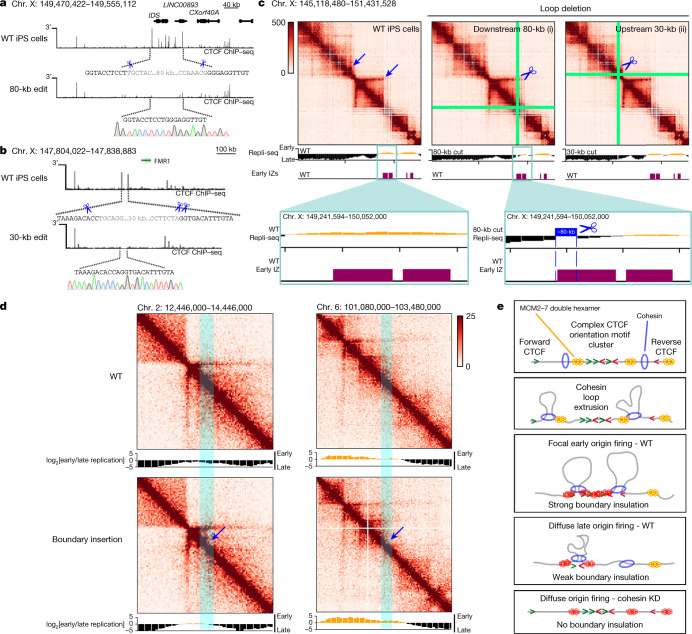
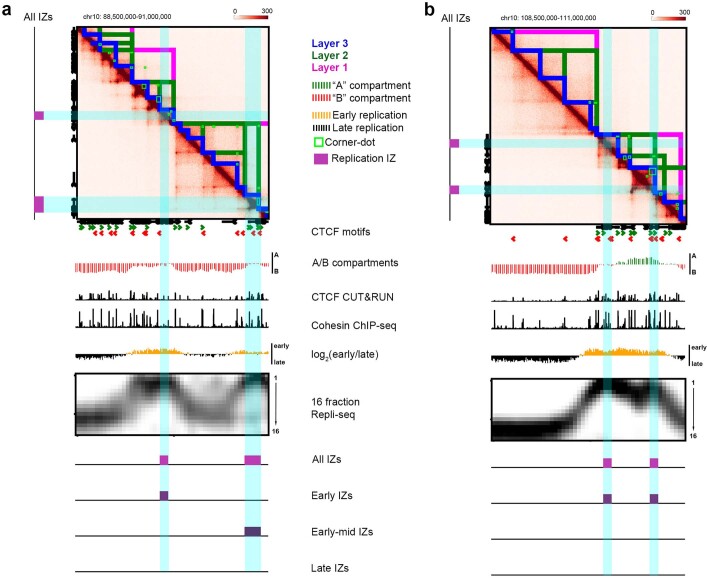
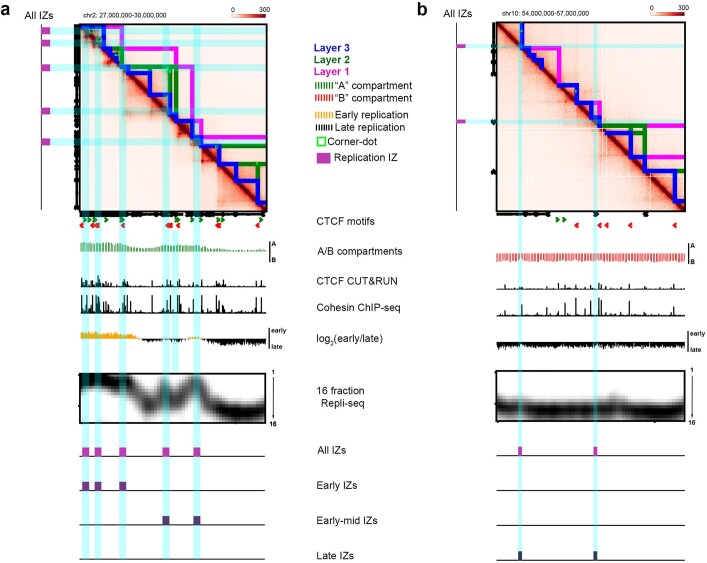
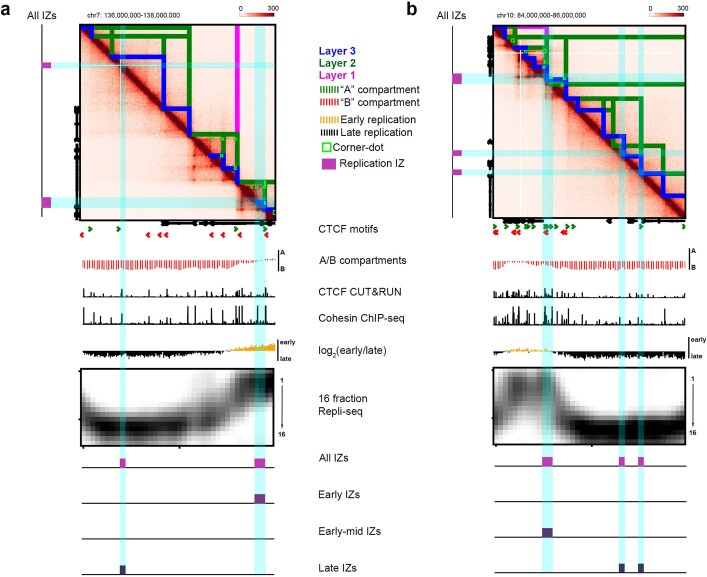
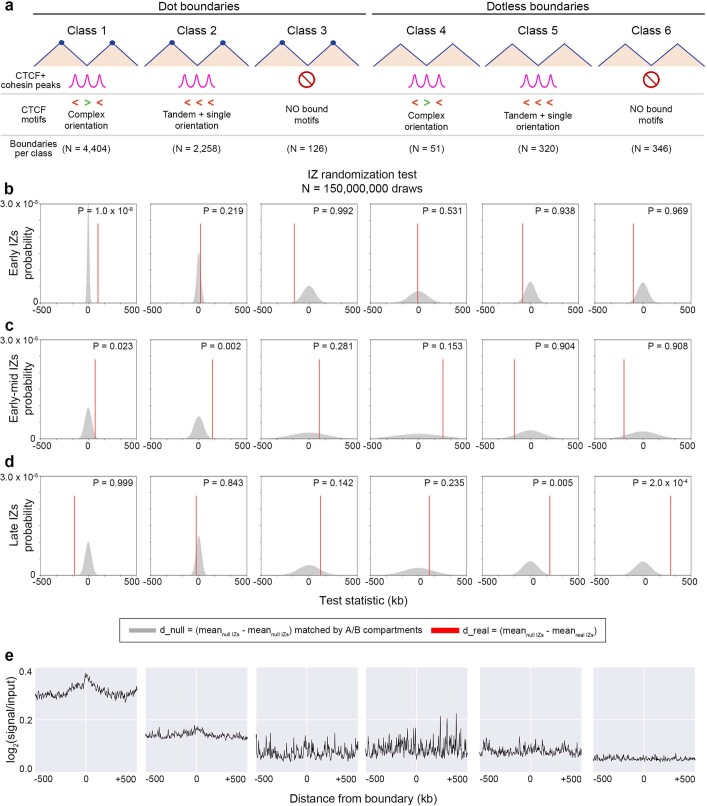
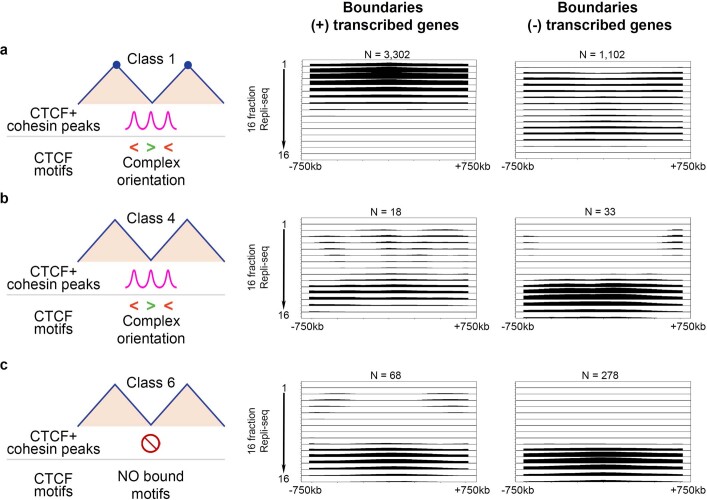
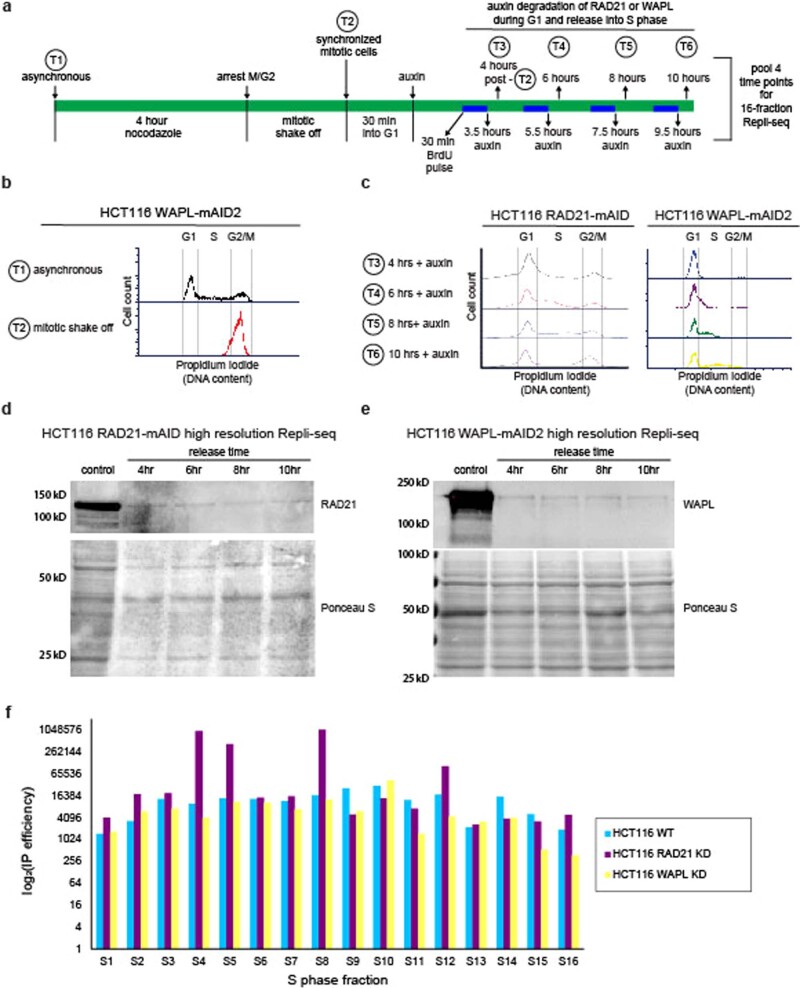
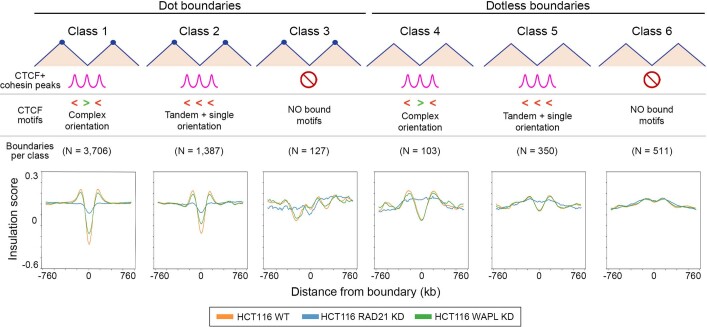
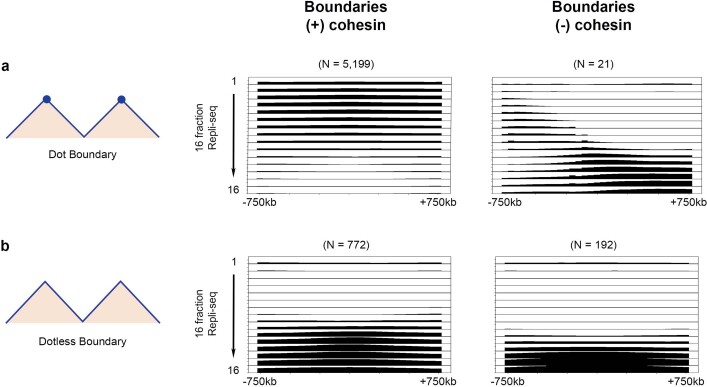
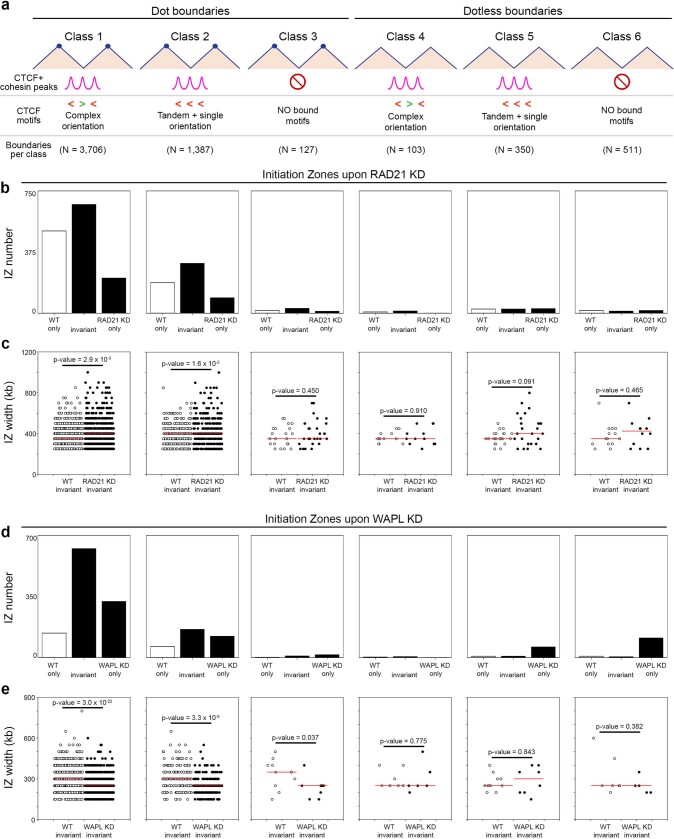
DNA replication occurs through an intricately regulated series of molecular events and is fundamental for genome stability1,2. At present, it is unknown how the locations of replication origins are determined in the human genome. Here we dissect the role of topologically associating domains (TADs)3-6, subTADs7 and loops8 in the positioning of replication initiation zones (IZs). We stratify TADs and subTADs by the presence of corner-dots indicative of loops and the orientation of CTCF motifs. We find that high-efficiency, early replicating IZs localize to boundaries between adjacent corner-dot TADs anchored by high-density arrays of divergently and convergently oriented CTCF motifs. By contrast, low-efficiency IZs localize to weaker dotless boundaries. Following ablation of cohesin-mediated loop extrusion during G1, high-efficiency IZs become diffuse and delocalized at boundaries with complex CTCF motif orientations. Moreover, G1 knockdown of the cohesin unloading factor WAPL results in gained long-range loops and narrowed localization of IZs at the same boundaries. Finally, targeted deletion or insertion of specific boundaries causes local replication timing shifts consistent with IZ loss or gain, respectively. Our data support a model in which cohesin-mediated loop extrusion and stalling at a subset of genetically encoded TAD and subTAD boundaries is an essential determinant of the locations of replication origins in human S phase.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
MeSH terms
Substances
Grants and funding
- R01 MH120269/MH/NIMH NIH HHS/United States
- UM1 HG011536/HG/NHGRI NIH HHS/United States
- R01 HG010658/HG/NHGRI NIH HHS/United States
- DP1 MH129957/MH/NIMH NIH HHS/United States
- U01 DA052715/DA/NIDA NIH HHS/United States
- U54 DK107965/DK/NIDDK NIH HHS/United States
- R37 MH120269/MH/NIMH NIH HHS/United States
- U01 DK127405/DK/NIDDK NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- U54 DK107980/DK/NIDDK NIH HHS/United States
- R01 GM083337/GM/NIGMS NIH HHS/United States
- U01 HL129998/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
