

This is a preprint.
RNA polymerase inaccuracy underlies SARS-CoV-2 variants and vaccine heterogeneity
- PMID: 35677076
- PMCID: PMC9176646
- DOI: 10.21203/rs.3.rs-1690086/v1
RNA polymerase inaccuracy underlies SARS-CoV-2 variants and vaccine heterogeneity
Update in
-
Targeted accurate RNA consensus sequencing (tARC-seq) reveals mechanisms of replication error affecting SARS-CoV-2 divergence.Nat Microbiol. 2024 May;9(5):1382-1392. doi: 10.1038/s41564-024-01655-4. Epub 2024 Apr 22. Nat Microbiol. 2024. PMID: 38649410 Free PMC article.
Abstract
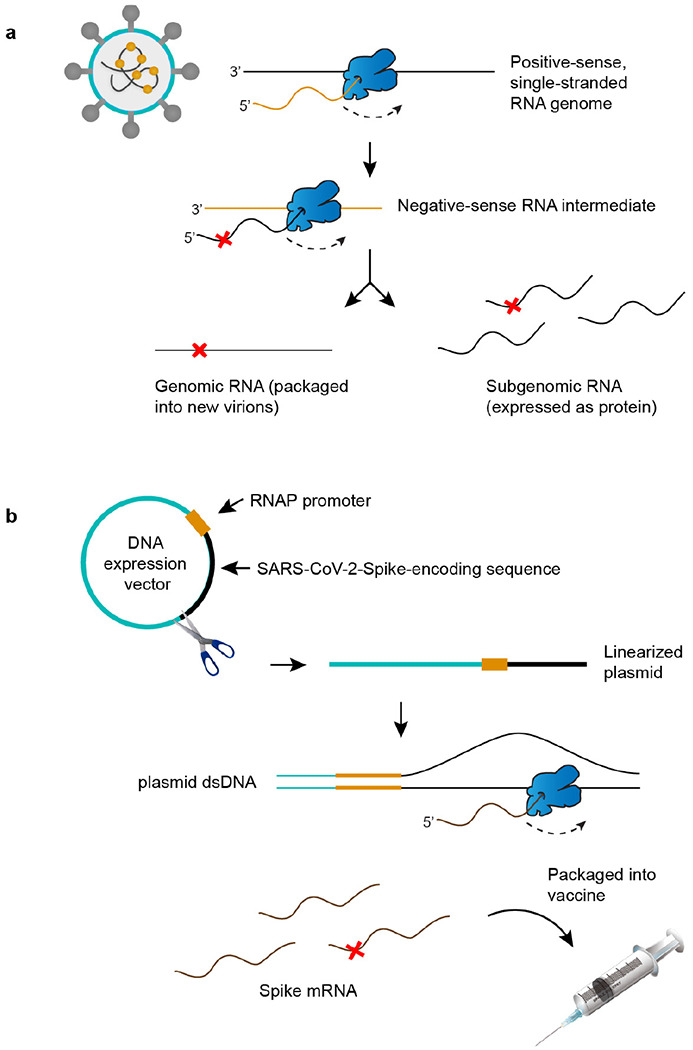
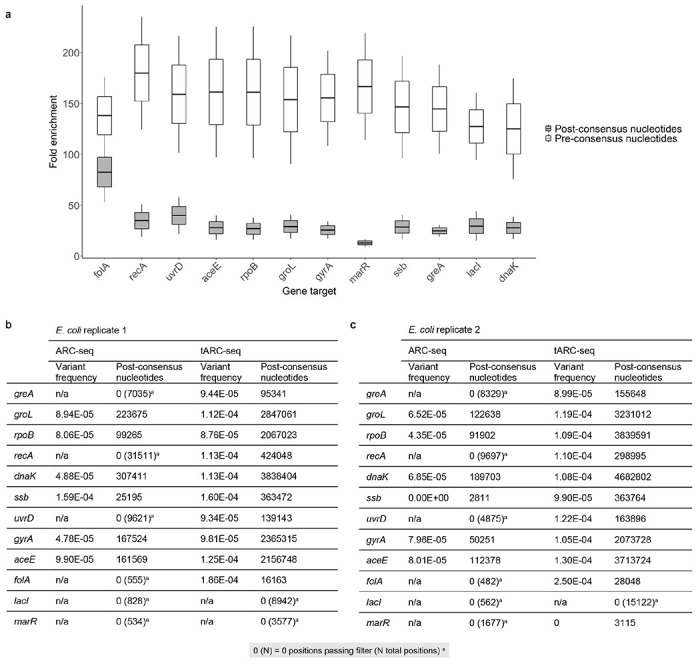
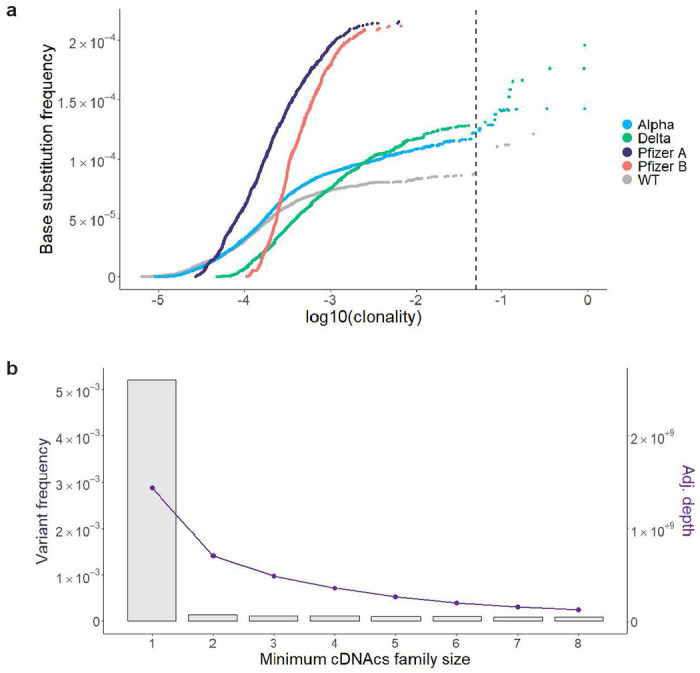
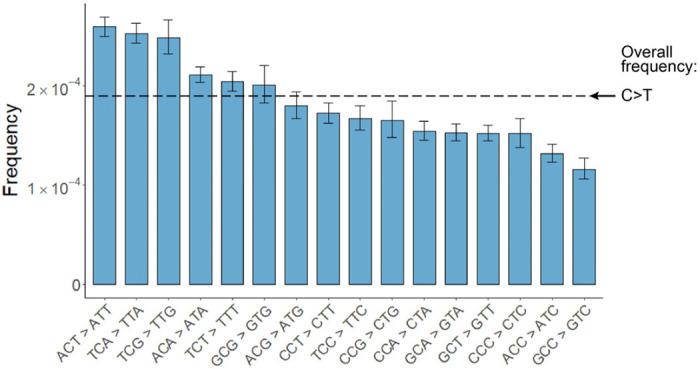
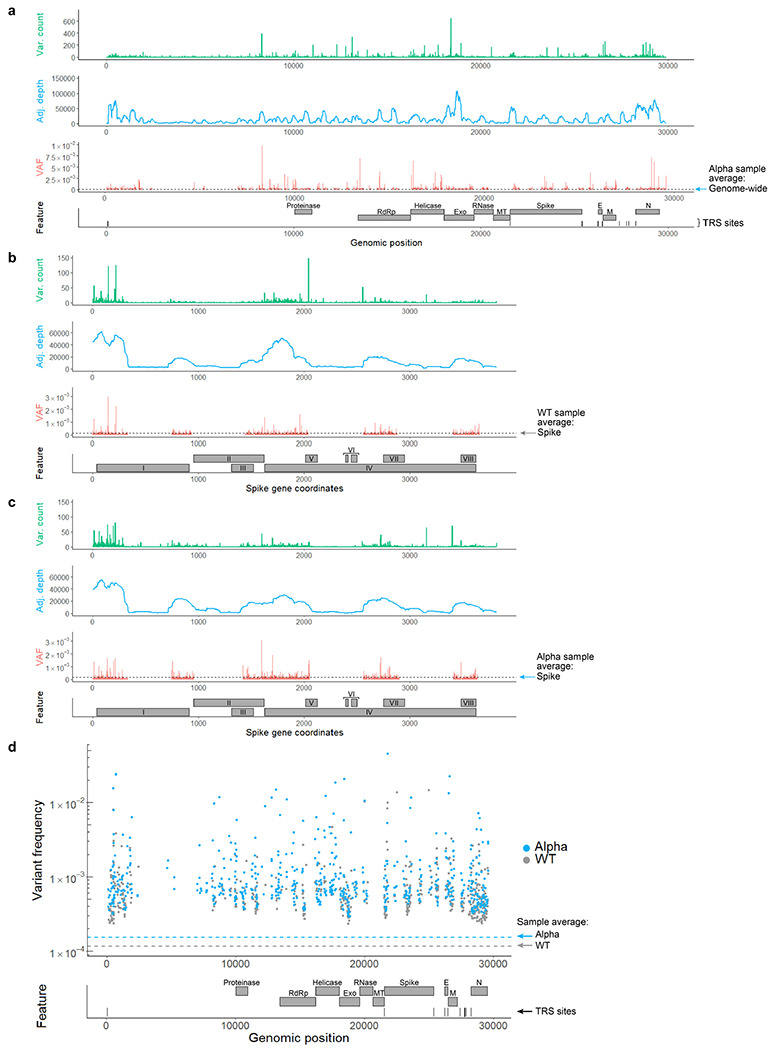
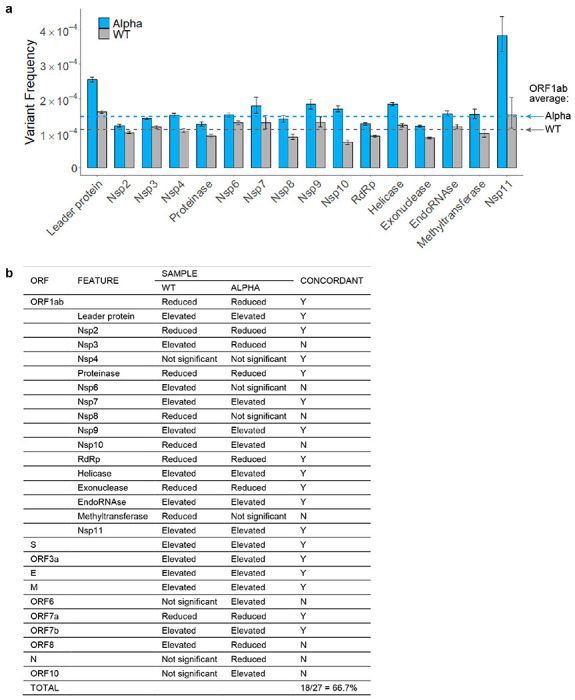
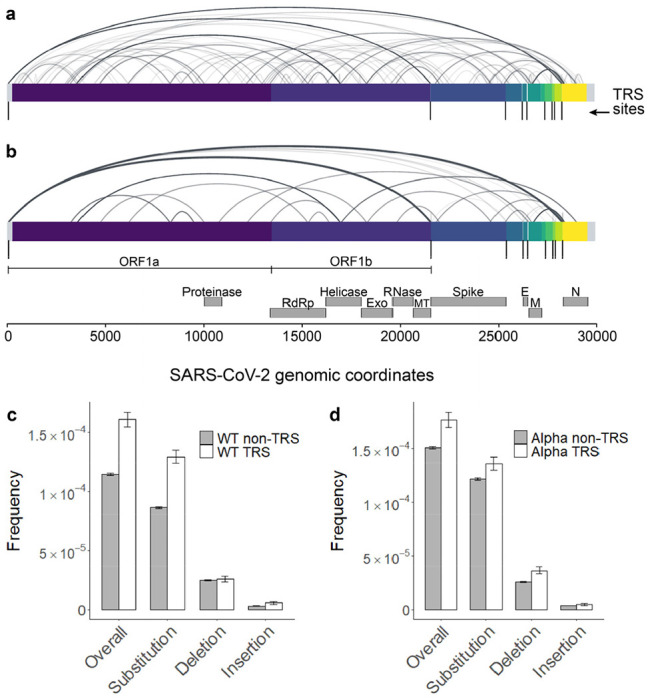
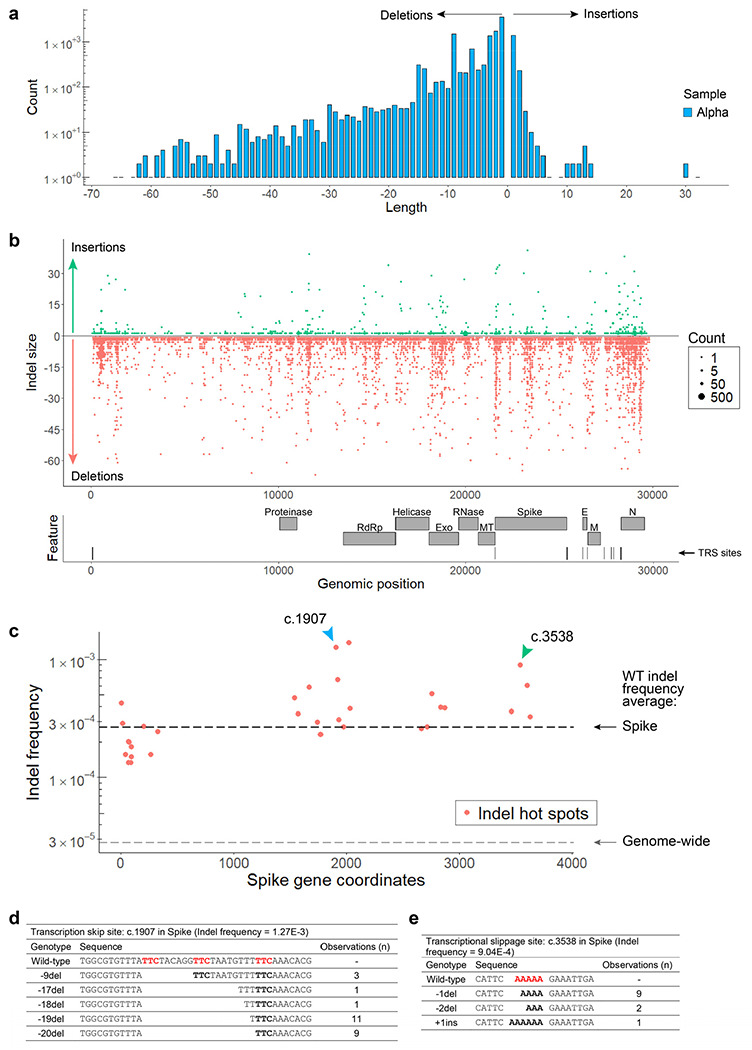
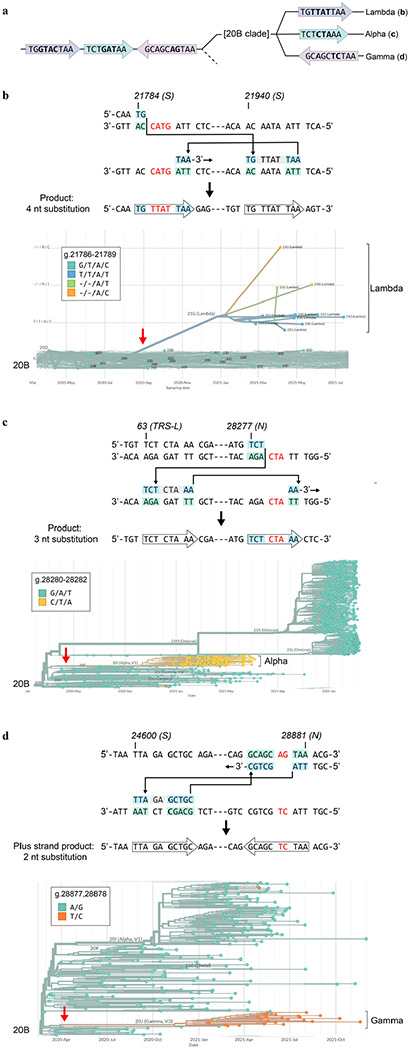
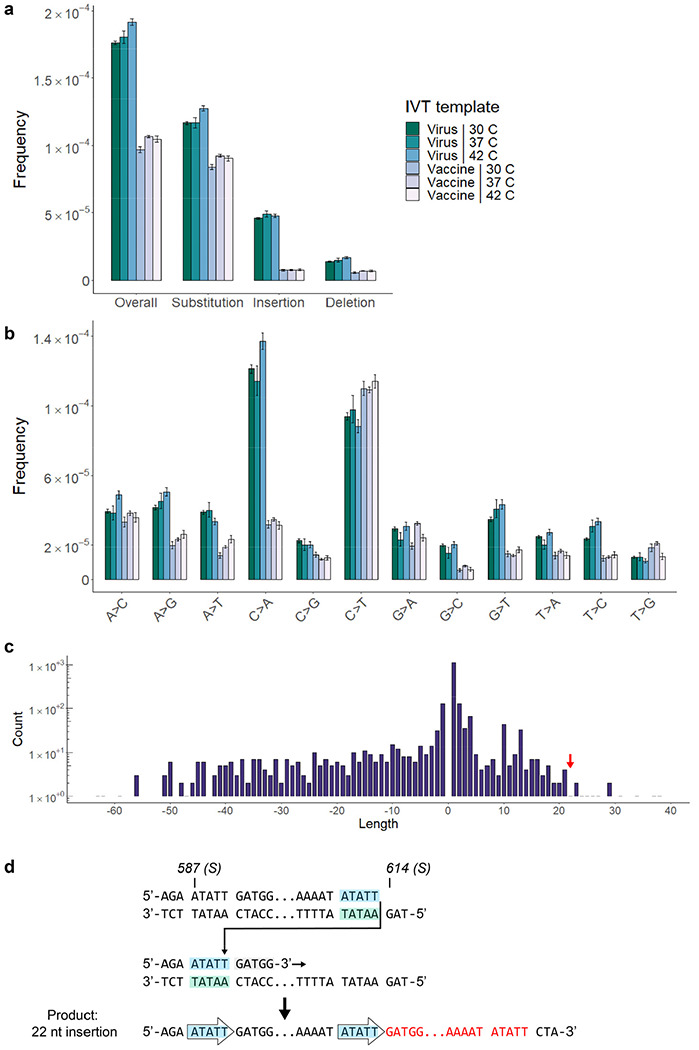
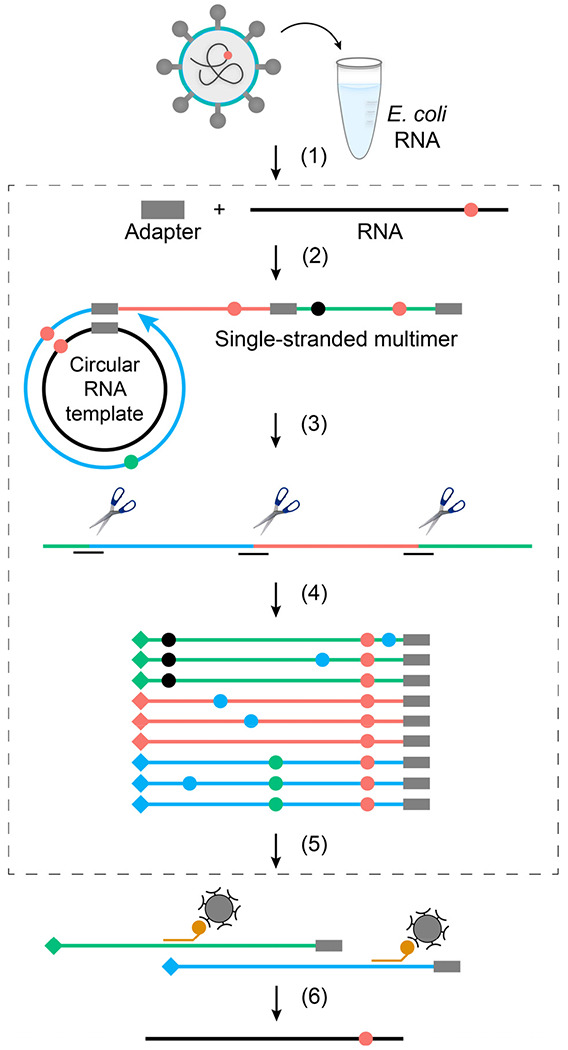
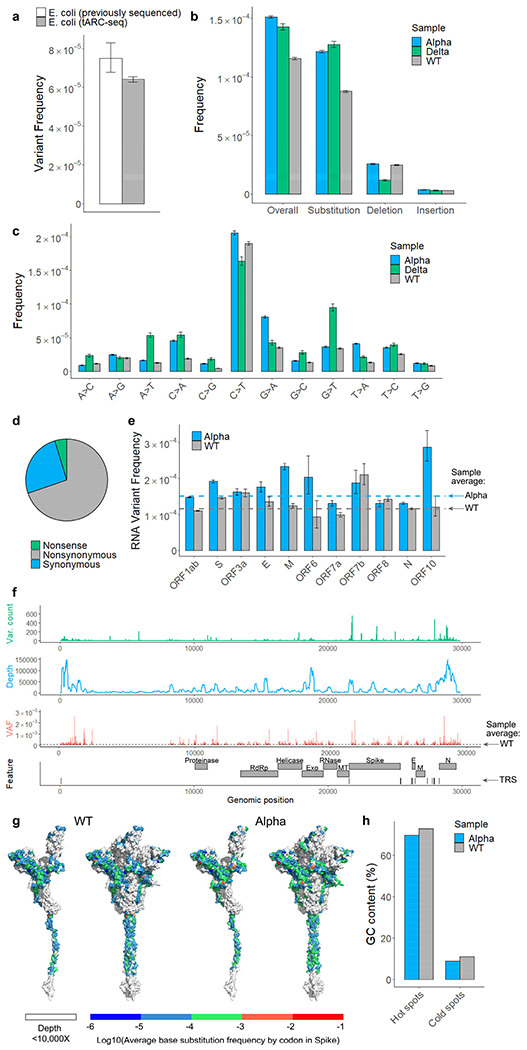
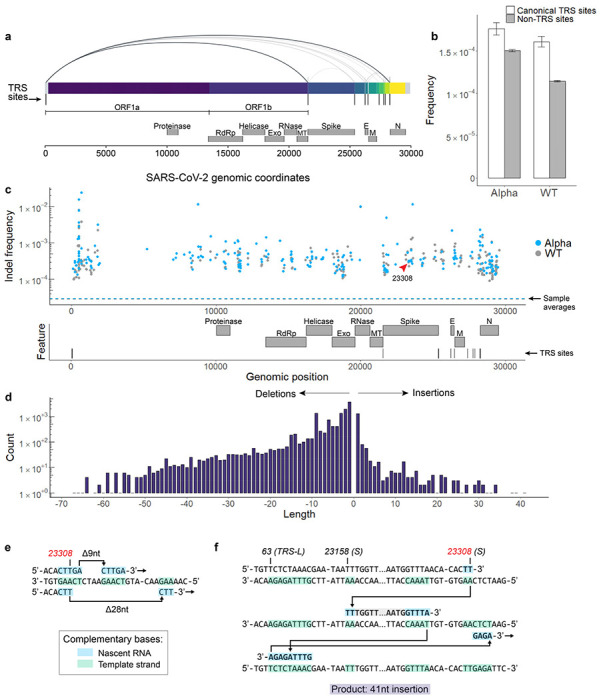
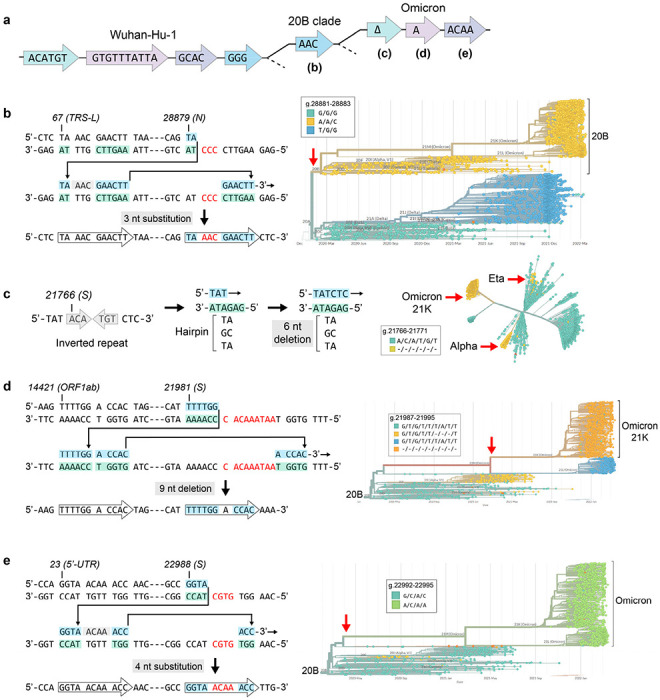
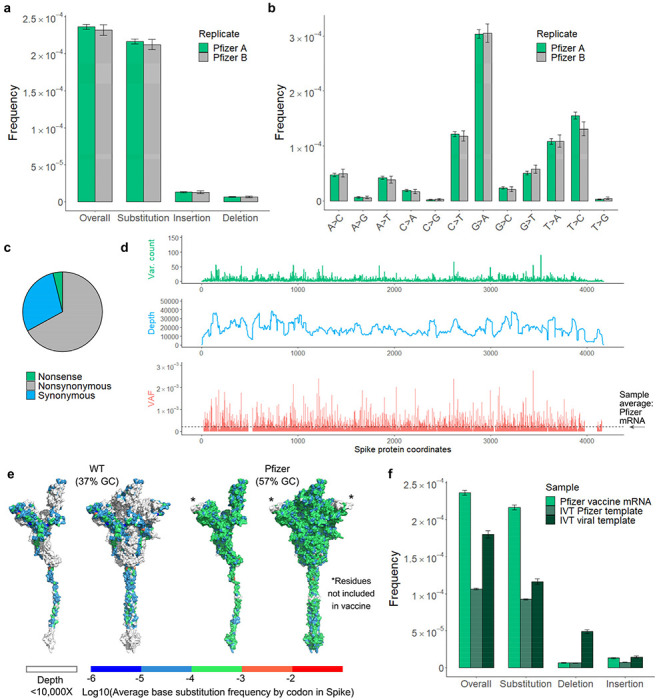
Both the SARS-CoV-2 virus and its mRNA vaccines depend on RNA polymerases (RNAP)1,2; however, these enzymes are inherently error-prone and can introduce variants into the RNA3. To understand SARS-CoV-2 evolution and vaccine efficacy, it is critical to identify the extent and distribution of errors introduced by the RNAPs involved in each process. Current methods lack the sensitivity and specificity to measure de novo RNA variants in low input samples like viral isolates3. Here, we determine the frequency and nature of RNA errors in both SARS-CoV-2 and its vaccine using a targeted Accurate RNA Consensus sequencing method (tARC-seq). We found that the viral RNA-dependent RNAP (RdRp) makes ~1 error every 10,000 nucleotides - higher than previous estimates4. We also observed that RNA variants are not randomly distributed across the genome but are associated with certain genomic features and genes, such as S (Spike). tARC-seq captured a number of large insertions, deletions and complex mutations that can be modeled through non-programmed RdRp template switching. This template switching feature of RdRp explains many key genetic changes observed during the evolution of different lineages worldwide, including Omicron. Further sequencing of the Pfizer-BioNTech COVID-19 vaccine revealed an RNA variant frequency of ~1 in 5,000, meaning most of the vaccine transcripts produced in vitro by T7 phage RNAP harbor a variant. These results demonstrate the extraordinary genetic diversity of viral populations and the heterogeneous nature of an mRNA vaccine fueled by RNAP inaccuracy. Along with functional studies and pandemic data, tARC-seq variant spectra can inform models to predict how SARS-CoV-2 may evolve. Finally, our results may help improve future vaccine development and study design as mRNA therapies continue to gain traction.
Conflict of interest statement
Competing interests The authors declare no competing interests.
Figures
Similar articles
-
Targeted accurate RNA consensus sequencing (tARC-seq) reveals mechanisms of replication error affecting SARS-CoV-2 divergence.Nat Microbiol. 2024 May;9(5):1382-1392. doi: 10.1038/s41564-024-01655-4. Epub 2024 Apr 22. Nat Microbiol. 2024. PMID: 38649410 Free PMC article.
-
The British variant of the new coronavirus-19 (Sars-Cov-2) should not create a vaccine problem.J Biol Regul Homeost Agents. 2021 Jan-Feb;35(1):1-4. doi: 10.23812/21-3-E. J Biol Regul Homeost Agents. 2021. PMID: 33377359
-
Comprehensive characterization of the antibody responses to SARS-CoV-2 Spike protein finds additional vaccine-induced epitopes beyond those for mild infection.Elife. 2022 Jan 24;11:e73490. doi: 10.7554/eLife.73490. Elife. 2022. PMID: 35072628 Free PMC article.
-
The COVID-19 pandemic: viral variants and vaccine efficacy.Crit Rev Clin Lab Sci. 2022 Jan;59(1):66-75. doi: 10.1080/10408363.2021.1979462. Epub 2021 Oct 1. Crit Rev Clin Lab Sci. 2022. PMID: 34598660 Review.
-
SARS-CoV-2 spike protein and RNA dependent RNA polymerase as targets for drug and vaccine development: A review.Biosaf Health. 2021 Oct;3(5):249-263. doi: 10.1016/j.bsheal.2021.07.003. Epub 2021 Jul 21. Biosaf Health. 2021. PMID: 34396086 Free PMC article. Review.
References
Main references
-
- Sahin U., Karikó K. & Türeci Ö. mRNA-based therapeutics--developing a new class of drugs. Nat Rev Drug Discov 13, 759–780 (2014). - PubMed
Methods references
-
- Rio D. C., Ares M., Hannon G. J. & Nilsen T. W. Purification of RNA Using TRIzol (TRI Reagent). Cold Spring Harb Protoc 2010, pdb.prot5439 (2010). - PubMed
-
- Reverse Engineering the source code of the BioNTech/Pfizer SARS-CoV-2 Vaccine. Bert Hubert’s writings https://berthub.eu/articles/posts/reverse-engineering-source-code-of-the... (2020).
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
