

Outcomes and genotype correlations in patients with mitochondrial trifunctional protein or isolated long chain 3-hydroxyacyl-CoA dehydrogenase deficiency enrolled in the IBEM-IS database
- PMID: 35677112
- PMCID: PMC9167967
- DOI: 10.1016/j.ymgmr.2022.100884
Outcomes and genotype correlations in patients with mitochondrial trifunctional protein or isolated long chain 3-hydroxyacyl-CoA dehydrogenase deficiency enrolled in the IBEM-IS database
Abstract
Purpose: Mitochondrial trifunctional protein deficiency (TFPD) and isolated long chain 3-hydroxyacyl-CoA dehydrogenase deficiency (LCHADD) are two related defects of fatty acid β -oxidation. While NBS has decreased mortality, morbidity remains significant. Additionally, the relationship of genotype to clinical outcome remains unclear. To better understand these issues, we collected natural history data for these conditions by reviewing seven years of retrospective data from 45 cases of TFPD or LCHADD in the Inborn Errors of Metabolism - Information System.
Methods: Available data included age at database entry, last datapoint, and development of various complications. Data were analyzed by clinical assigned diagnosis (LCHADD or TFPD), subdivided by method of ascertainment (newborn screening-NBS, or other than by newborn screening-NNBS), then re-analyzed based on four genotype groups: homozygous c.1528GC (p.E510Q) (common LCHAD variant); heterozygous c.1528GC (p.E510Q), other HADHA variants; and HADHB variants.
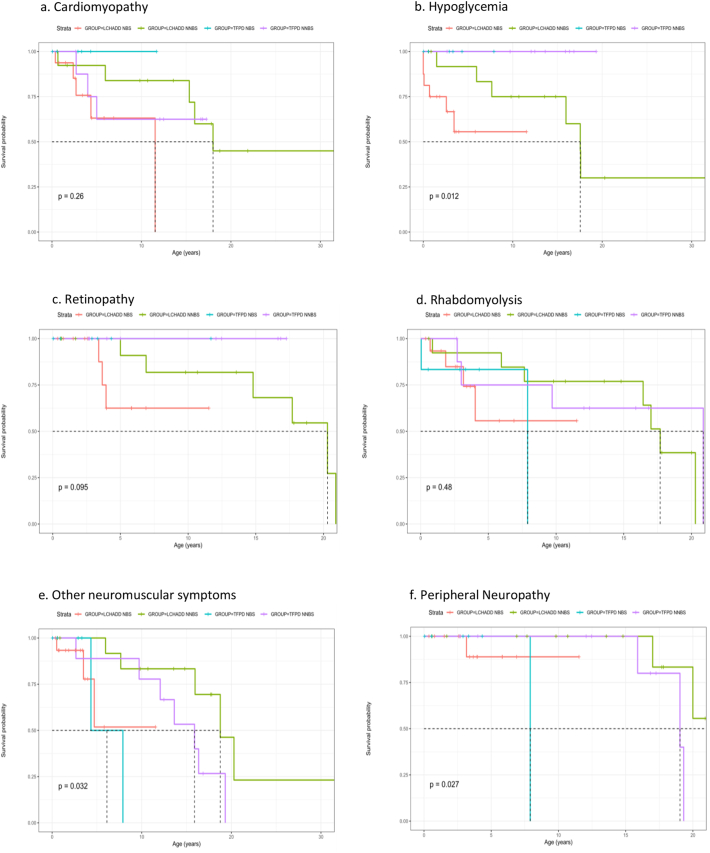
Results: Forty-five patients from birth to 34 years of age were analyzed by assigned diagnosis (30 LCHADD and 15 TFPD) and method of ascertainment. Thirty had further analysis by genotype (22 biallelic HADHA variants and 8 biallelic HADHB variants). With regards to maternal complications, retinopathy, cardiomyopathy and hypoglycemia, patients with biallelic HADHA variants (with or without the common LCHAD variant) manifest a traditional LCHADD phenotype, while those with HADHB gene variants more commonly reported neuromusculoskeletal type TFPD phenotype. While retinopathy, rhabdomyolysis and peripheral neuropathy tended to present later in childhood, many features including initial report of cardiomyopathy and hypoglycemia presented across a wide age spectrum.
Conclusion: This study demonstrates the utility of genotypic confirmation of patients identified with LCHADD/TFPD as variants in the HADHA and HADHB genes lead to different symptom profiles. In our data, biallelic HAHDA variants conferred a LCHADD phenotype, regardless of the presence of the common LCHAD variant.
Keywords: Fatty acid oxidation disorders; Genetics; Inborn errors of metabolism; LCHAD; MTFP; Mitochondrial trifunctional protein; Pediatrics; TFP; Trifunctional protein.
© 2022 The Authors.
Similar articles
-
Genetic, biochemical, and clinical spectrum of patients with mitochondrial trifunctional protein deficiency identified after the introduction of newborn screening in the Netherlands.J Inherit Metab Dis. 2022 Jul;45(4):804-818. doi: 10.1002/jimd.12502. Epub 2022 Apr 19. J Inherit Metab Dis. 2022. PMID: 35383965 Free PMC article.
-
Plasma Metabolomics, Lipidomics, and Acylcarnitines Are Associated With Vision and Genotype but Not With Dietary Intake in Long-Chain 3-Hydroxyacyl-CoA Dehydrogenase Deficiency (LCHADD).J Inherit Metab Dis. 2025 Jul;48(4):e70060. doi: 10.1002/jimd.70060. J Inherit Metab Dis. 2025. PMID: 40635623
-
Observations regarding retinopathy in mitochondrial trifunctional protein deficiencies.Mol Genet Metab. 2012 May;106(1):18-24. doi: 10.1016/j.ymgme.2012.02.015. Epub 2012 Mar 8. Mol Genet Metab. 2012. PMID: 22459206 Free PMC article. Review.
-
Characterization of Chorioretinopathy Associated with Mitochondrial Trifunctional Protein Disorders: Long-Term Follow-up of 21 Cases.Ophthalmology. 2016 Oct;123(10):2183-95. doi: 10.1016/j.ophtha.2016.06.048. Epub 2016 Aug 2. Ophthalmology. 2016. PMID: 27491397 Free PMC article.
-
Sudden Death of a Four-Day-Old Newborn Due to Mitochondrial Trifunctional Protein/Long-Chain 3-Hydroxyacyl-CoA Dehydrogenase Deficiencies and a Systematic Literature Review of Early Deaths of Neonates with Fatty Acid Oxidation Disorders.Int J Neonatal Screen. 2025 Jan 26;11(1):9. doi: 10.3390/ijns11010009. Int J Neonatal Screen. 2025. PMID: 39982343 Free PMC article. Review.
Cited by
-
Neurological outcome in long-chain hydroxy fatty acid oxidation disorders.Ann Clin Transl Neurol. 2024 Apr;11(4):883-898. doi: 10.1002/acn3.52002. Epub 2024 Jan 23. Ann Clin Transl Neurol. 2024. PMID: 38263760 Free PMC article.
-
HADHA Regulates Respiratory Complex Assembly and Couples FAO and OXPHOS.Adv Sci (Weinh). 2024 Dec;11(47):e2405147. doi: 10.1002/advs.202405147. Epub 2024 Nov 3. Adv Sci (Weinh). 2024. PMID: 39488787 Free PMC article.
-
A proposal for an updated staging system for LCHADD retinopathy.Ophthalmic Genet. 2024 Apr;45(2):140-146. doi: 10.1080/13816810.2024.2303682. Epub 2024 Jan 30. Ophthalmic Genet. 2024. PMID: 38288966 Free PMC article.
-
Outcomes of mitochondrial long chain fatty acid oxidation and carnitine defects from a single center metabolic genetics clinic.Orphanet J Rare Dis. 2022 Sep 15;17(1):360. doi: 10.1186/s13023-022-02512-5. Orphanet J Rare Dis. 2022. PMID: 36109795 Free PMC article. Review.
-
Diagnostic challenges and outcome of fatty acid oxidation defects in a tertiary care center in Lebanon.Orphanet J Rare Dis. 2024 Aug 29;19(1):315. doi: 10.1186/s13023-024-03325-4. Orphanet J Rare Dis. 2024. PMID: 39210374 Free PMC article.
References
-
- Watson M.S., Mann M.Y., Lloyd-puryear M.A., Rinaldo P. Executive summary. Genet. Med. 2006;8:1–11. - PubMed
-
- Houten S.M., Violante S., Ventura F.V., Wanders R.J.A. The biochemistry and physiology of mitochondrial fatty acid β-oxidation and its genetic disorders. Annu. Rev. Physiol. 2016;78:23–44. - PubMed
-
- Ibdah J.A., Dasouki M.J., Strauss A.W. Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: variable expressivity of maternal illness during pregnancy and unusual presentation with infantile cholestasis and hypocalcaemia. J. Inherit. Metab. Dis. 1999;22:811–814. - PubMed
-
- Strauss A.W., et al. Inherited long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency and a fetal-maternal interaction cause maternal liver disease and other pregnancy complications. Semin. Perinatol. 1999;23:100–112. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
