

tRNA dysregulation and disease
- PMID: 35681060
- PMCID: PMC11170316
- DOI: 10.1038/s41576-022-00501-9
tRNA dysregulation and disease
Abstract
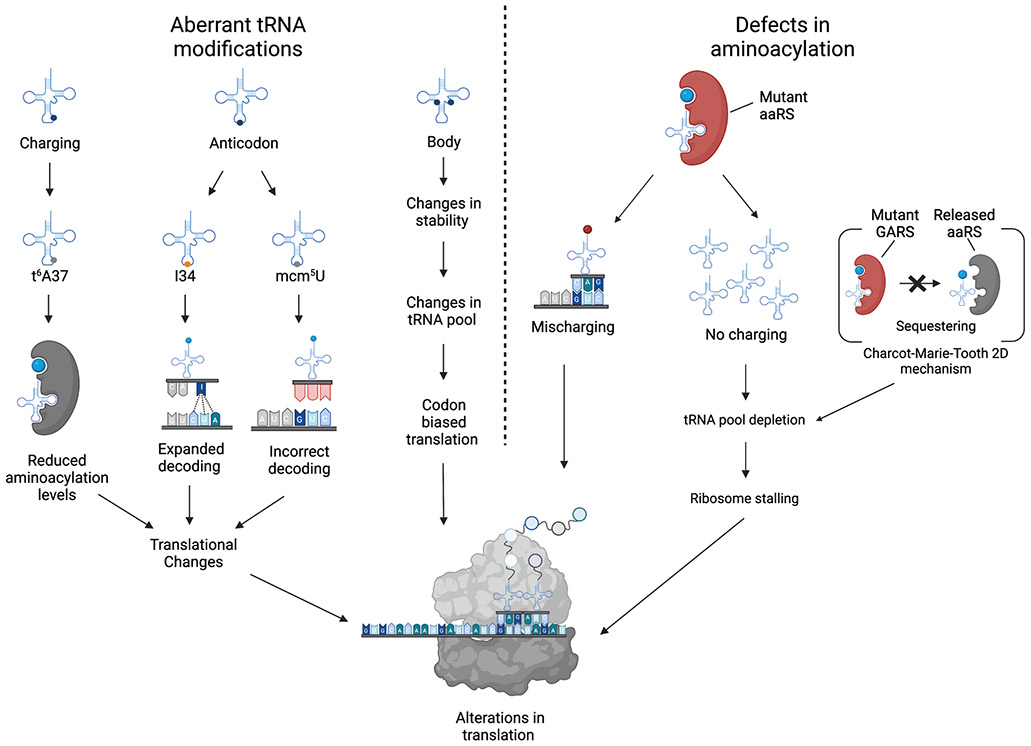
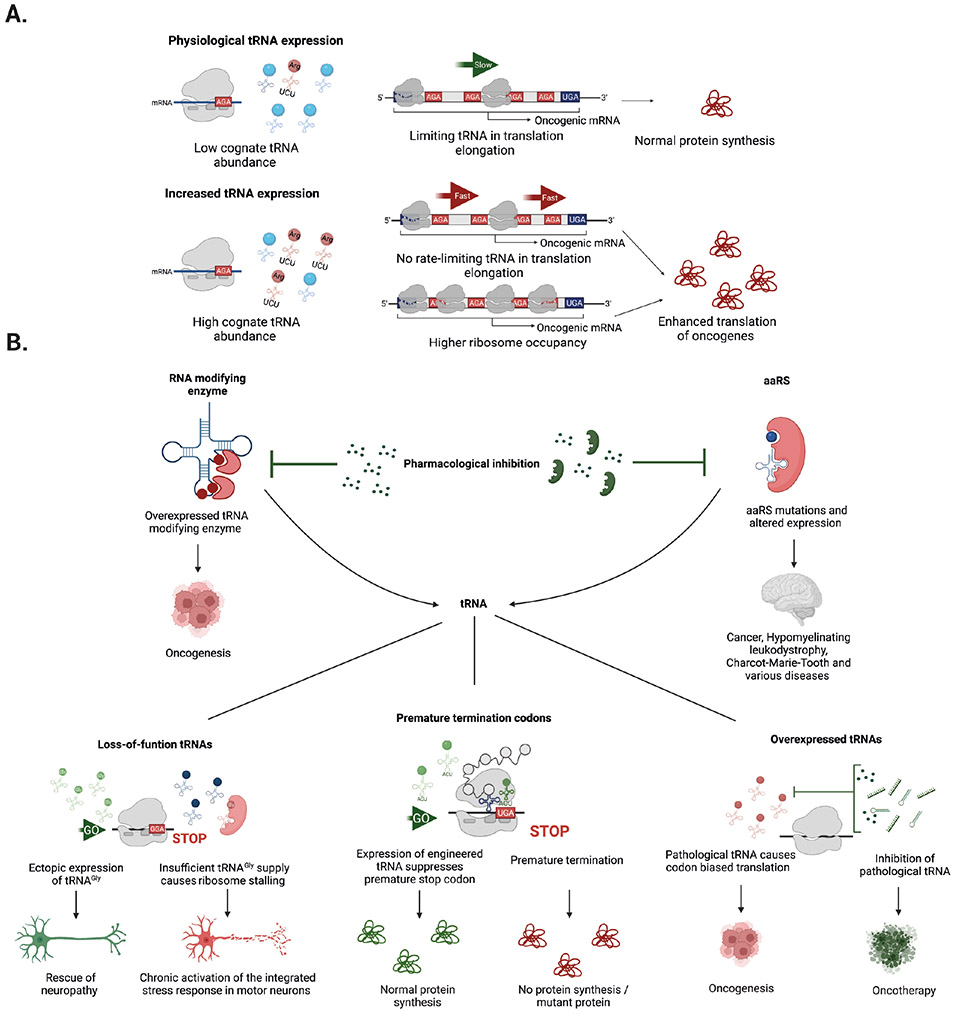
tRNAs are key adaptor molecules that decipher the genetic code during translation of mRNAs in protein synthesis. In contrast to the traditional view of tRNAs as ubiquitously expressed housekeeping molecules, awareness is now growing that tRNA-encoding genes display tissue-specific and cell type-specific patterns of expression, and that tRNA gene expression and function are both dynamically regulated by post-transcriptional RNA modifications. Moreover, dysregulation of tRNAs, mediated by alterations in either their abundance or function, can have deleterious consequences that contribute to several distinct human diseases, including neurological disorders and cancer. Accumulating evidence shows that reprogramming of mRNA translation through altered tRNA activity can drive pathological processes in a codon-dependent manner. This Review considers the emerging evidence in support of the precise control of functional tRNA levels as an important regulatory mechanism that coordinates mRNA translation and protein expression in physiological cell homeostasis, and highlights key examples of human diseases that are linked directly to tRNA dysregulation.
© 2022. Springer Nature Limited.
Figures
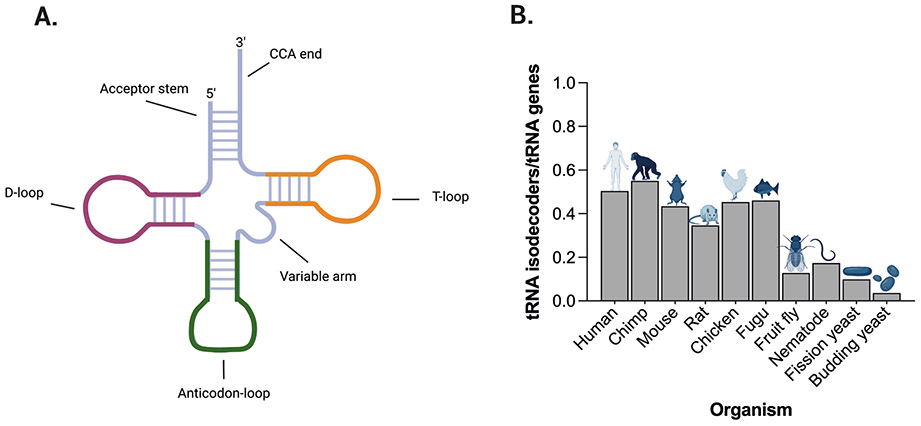
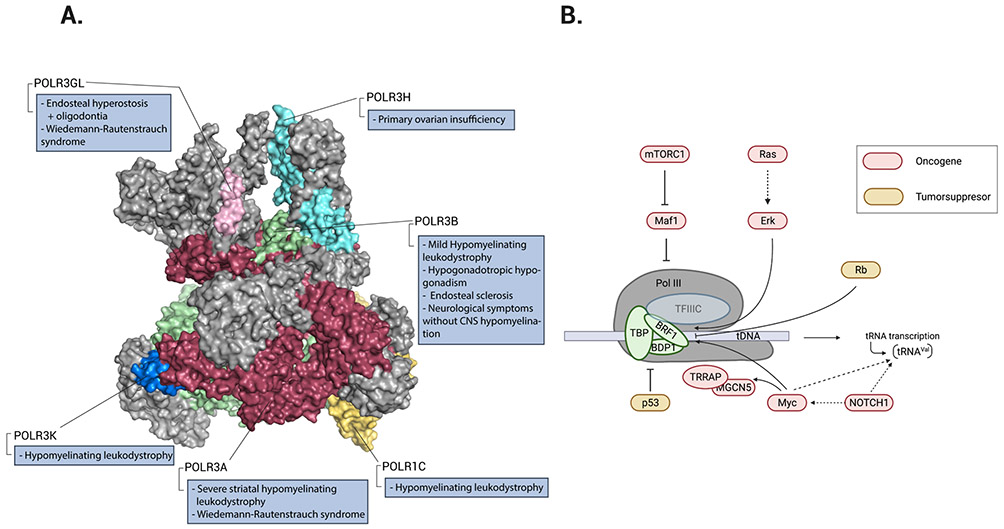
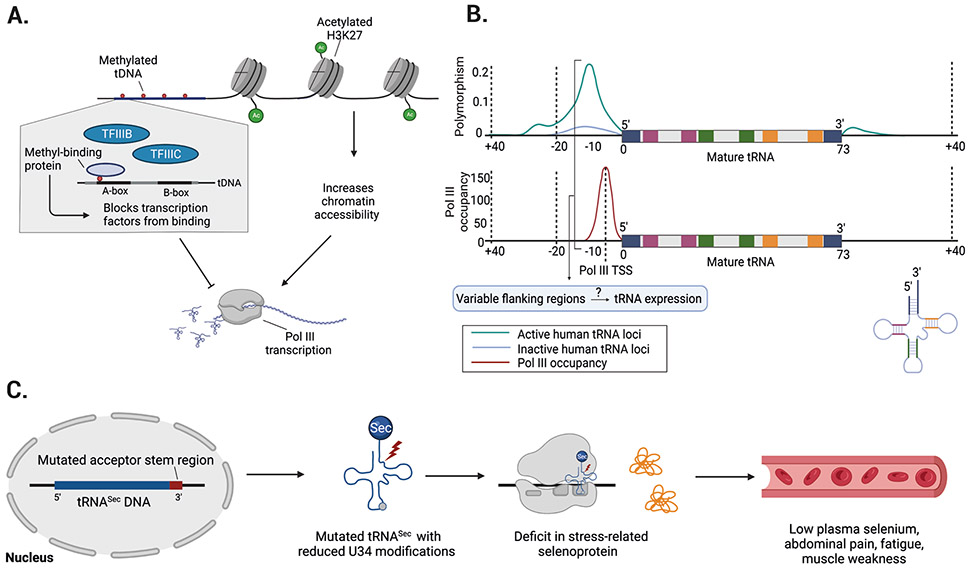
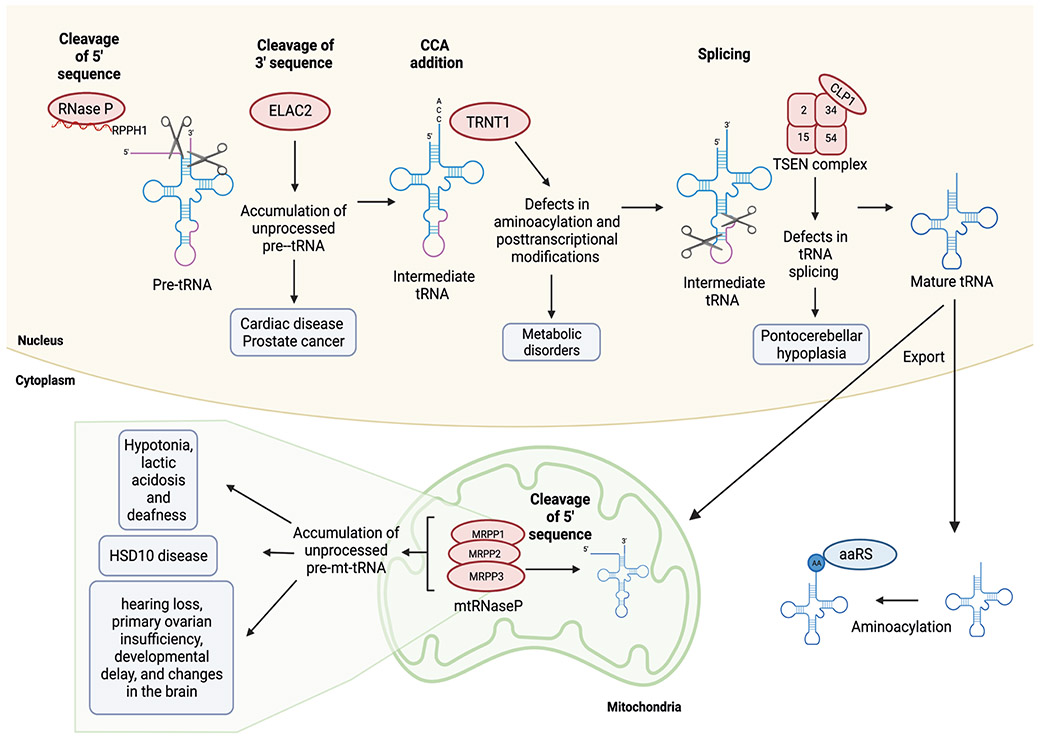
References
-
-
Hoagland MB, Stephenson ML, Scott JF, Hecht LI & Zamecick PC A soluble ribonucleic acid intermediate in protein synthesis. J. Biol. Chem 231, 241–257 (1958).
This paper described the discovery of tRNAs.
-
-
-
Holley RW et al. Structure of a ribonucleic acid. Science 147, 1462–1465 (1965).
This paper described the structure of tRNAs and the finding of a modified base (inosine).
-
-
-
Crick FHC Codon-anticodon pairing: the wobble hypothesis. J. Mol. Biol 19, 548–555 (1966).
This paper described the possibility of non-canonical (wobble) pairing of the anticodon loop with its cognate codon sequence on mRNAs.
-
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
