

Expanding the Molecular Spectrum of ANKRD11 Gene Defects in 33 Patients with a Clinical Presentation of KBG Syndrome
- PMID: 35682590
- PMCID: PMC9180463
- DOI: 10.3390/ijms23115912
Expanding the Molecular Spectrum of ANKRD11 Gene Defects in 33 Patients with a Clinical Presentation of KBG Syndrome
Abstract
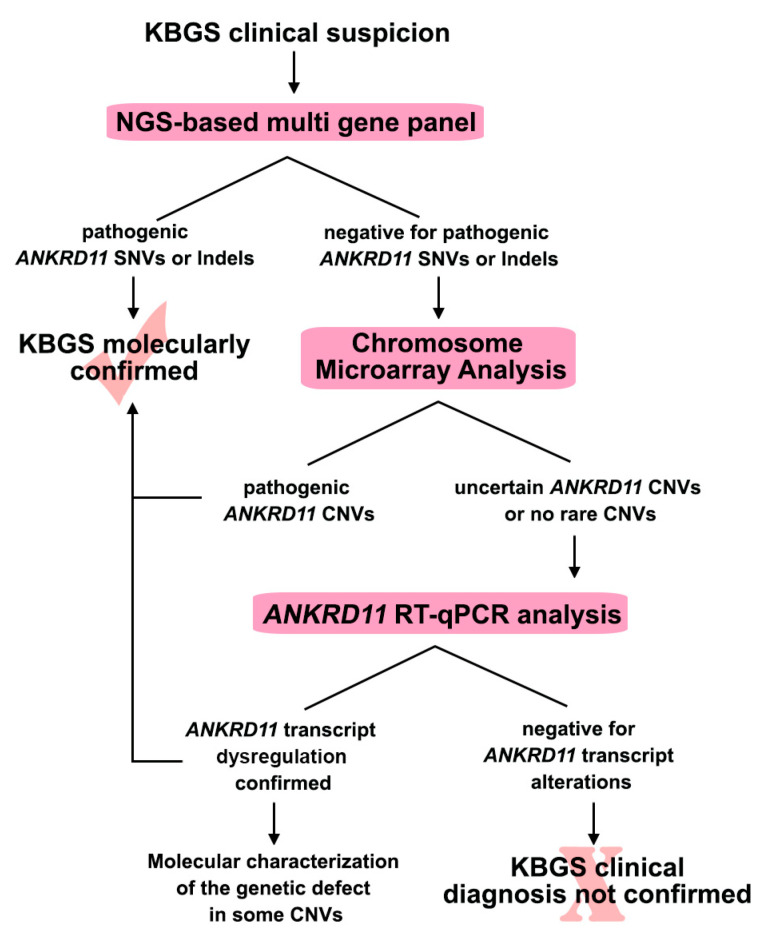
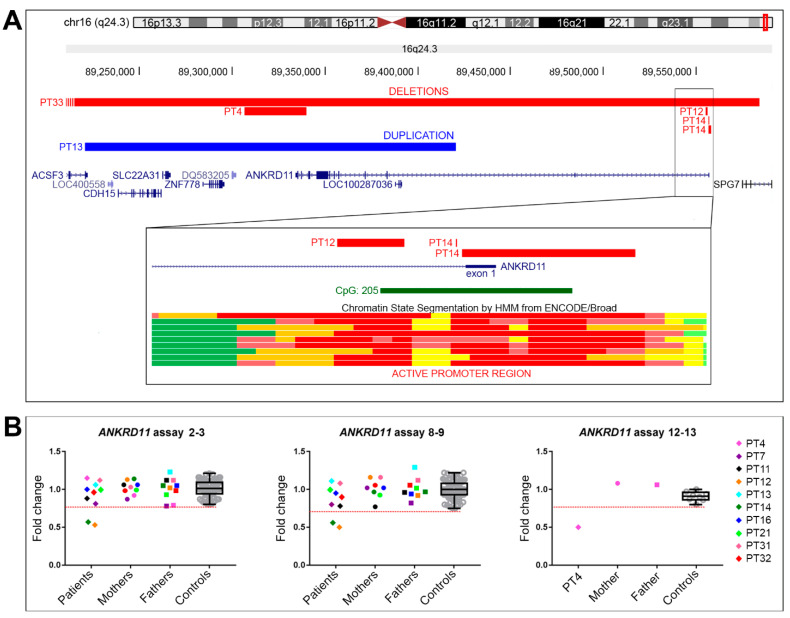
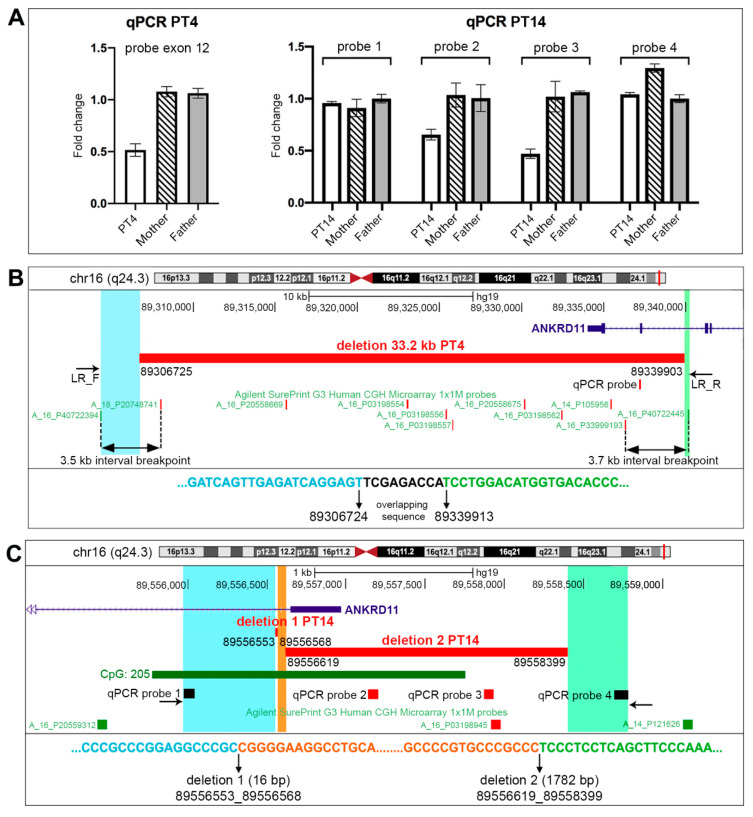
KBG syndrome (KBGS) is a neurodevelopmental disorder caused by the Ankyrin Repeat Domain 11 (ANKRD11) haploinsufficiency. Here, we report the molecular investigations performed on a cohort of 33 individuals with KBGS clinical suspicion. By using a multi-testing genomic approach, including gene sequencing, Chromosome Microarray Analysis (CMA), and RT-qPCR gene expression assay, we searched for pathogenic alterations in ANKRD11. A molecular diagnosis was obtained in 22 out of 33 patients (67%). ANKRD11 sequencing disclosed pathogenic or likely pathogenic variants in 18 out of 33 patients. CMA identified one full and one terminal ANKRD11 pathogenic deletions, and one partial duplication and one intronic microdeletion, with both possibly being pathogenic. The pathogenic effect was established by RT-qPCR, which confirmed ANKRD11 haploinsufficiency only for the three deletions. Moreover, RT-qPCR applied to six molecularly unsolved KBGS patients identified gene downregulation in a clinically typical patient with previous negative tests, and further molecular investigations revealed a cryptic deletion involving the gene promoter. In conclusion, ANKRD11 pathogenic variants could also involve the regulatory regions of the gene. Moreover, the application of a multi-test approach along with the innovative use of RT-qPCR improved the diagnostic yield in KBGS suspected patients.
Keywords: ANKRD11 gene expression analysis; ANKRD11 variations; KBG syndrome; diagnostic flow chart.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Herrmann J., Pallister P.D., Tiddy W., Opitz J.M. The KBG syndrome-a syndrome of short stature, characteristic facies, mental retardation, macrodontia and skeletal anomalies. Birth Defects. Orig. Artic. Ser. 1975;11:7–18. - PubMed
-
- Sirmaci A., Spiliopoulos M., Brancati F., Powell E., Duman D., Abrams A., Bademci G., Agolini E., Guo S., Konuk B., et al. Mutations in ANKRD11 cause KBG syndrome, characterized by intellectual disability, skeletal malformations, and macrodontia. Am. J. Hum. Genet. 2011;89:289–294. doi: 10.1016/j.ajhg.2011.06.007. - DOI - PMC - PubMed
