

Molecular Glues: Capable Protein-Binding Small Molecules That Can Change Protein-Protein Interactions and Interactomes for the Potential Treatment of Human Cancer and Neurodegenerative Diseases
- PMID: 35682885
- PMCID: PMC9181451
- DOI: 10.3390/ijms23116206
Molecular Glues: Capable Protein-Binding Small Molecules That Can Change Protein-Protein Interactions and Interactomes for the Potential Treatment of Human Cancer and Neurodegenerative Diseases
Abstract
Molecular glue (MG) compounds are a type of unique small molecule that can change the protein-protein interactions (PPIs) and interactomes by degrading, stabilizing, or activating the target protein after their binging. These small-molecule MGs are gradually being recognized for their potential application in treating human diseases, including cancer. Evidence suggests that small-molecule MG compounds could essentially target any proteins, which play critical roles in human disease etiology, where many of these protein targets were previously considered undruggable. Intriguingly, most MG compounds with high efficacy for cancer treatment can glue on and control multiple key protein targets. On the other hand, a single key protein target can also be glued by multiple MG compounds with distinct chemical structures. The high flexibility of MG-protein interaction profiles provides rich soil for the growth and development of small-molecule MG compounds that can be used as molecular tools to assist in unraveling disease mechanisms, and they can also facilitate drug development for the treatment of human disease, especially human cancer. In this review, we elucidate this concept by using various types of small-molecule MG compounds and their corresponding protein targets that have been documented in the literature.
Keywords: E3 ligase protein complex; cancer; human disease; molecular glue (MG); neurodegenerative disease; protein degradation; protein ubiquitination; small-molecule compounds.
Conflict of interest statement
FL118 and FL118 core structure-based analogs will be further developed in Canget BioTekpharma LLC (
Figures
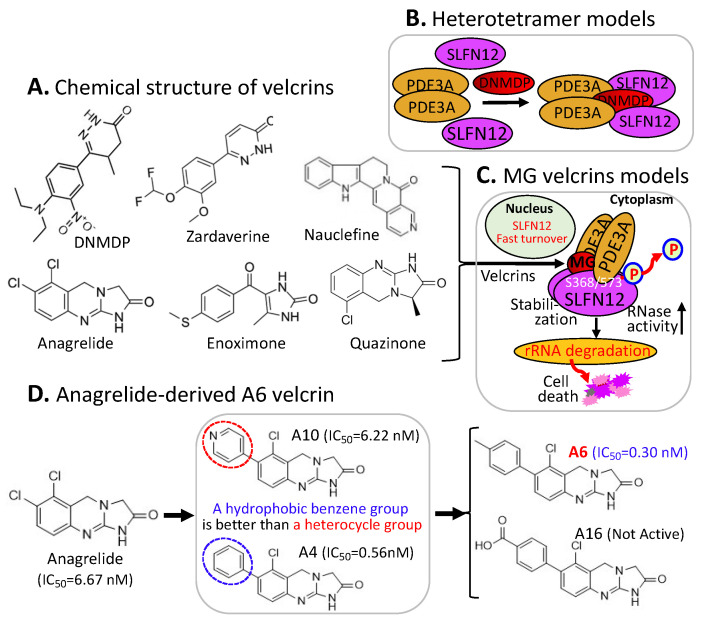
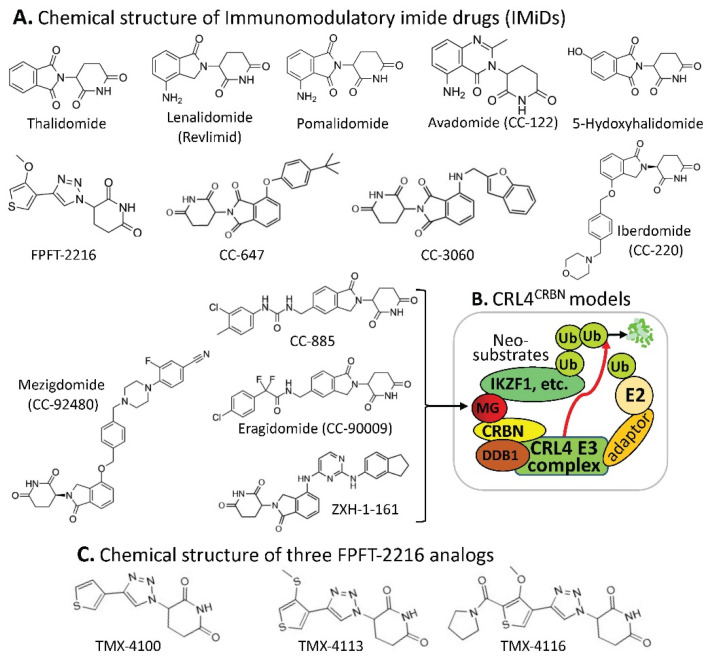
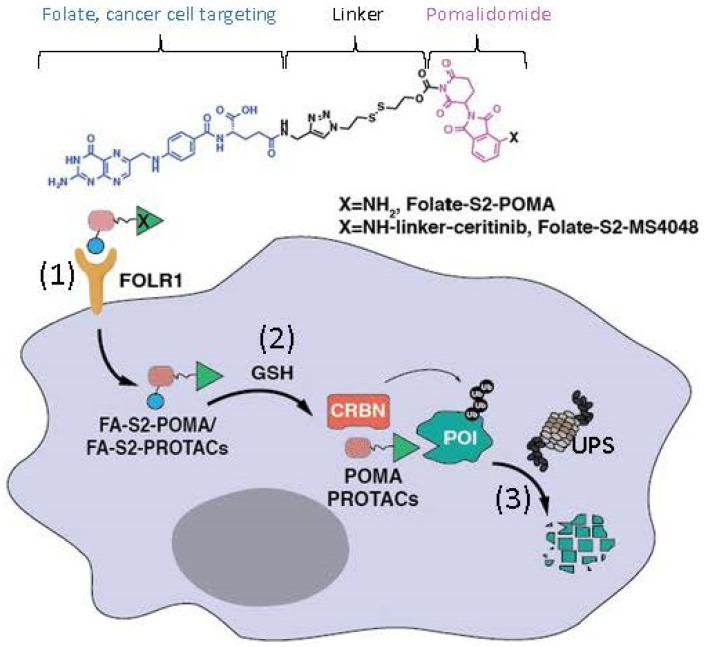
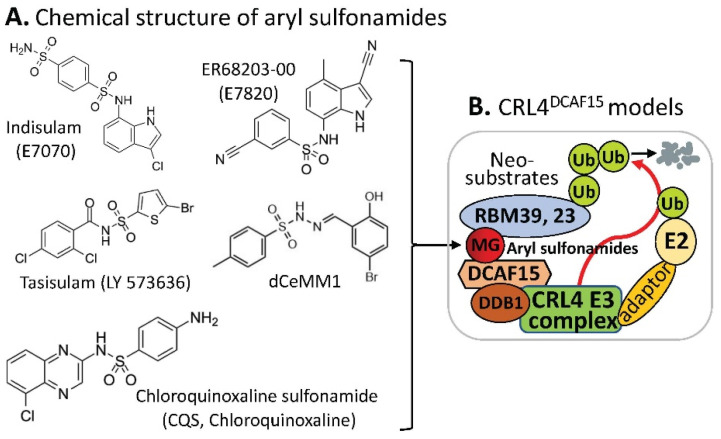
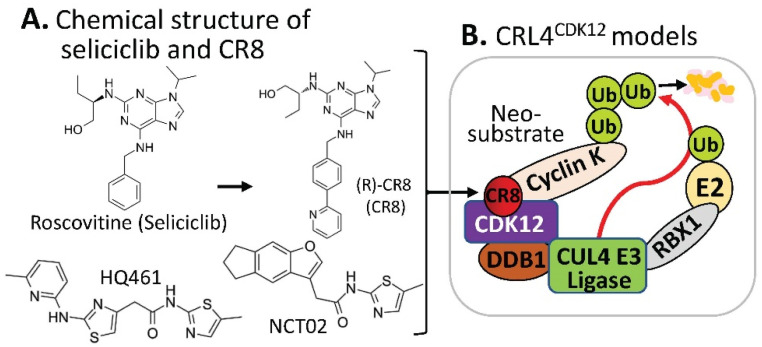
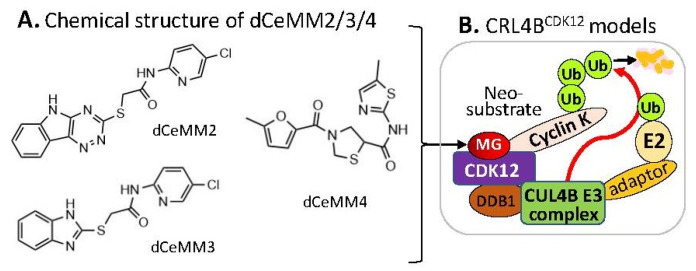
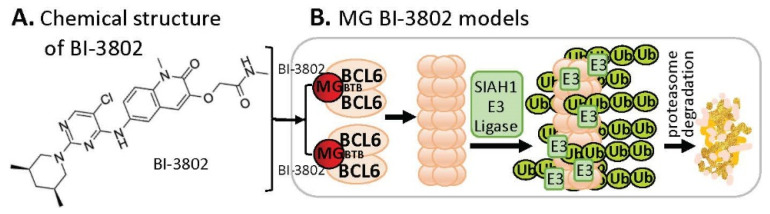
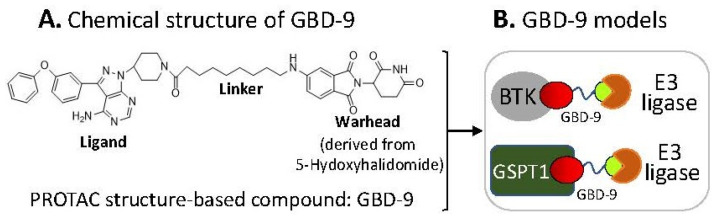
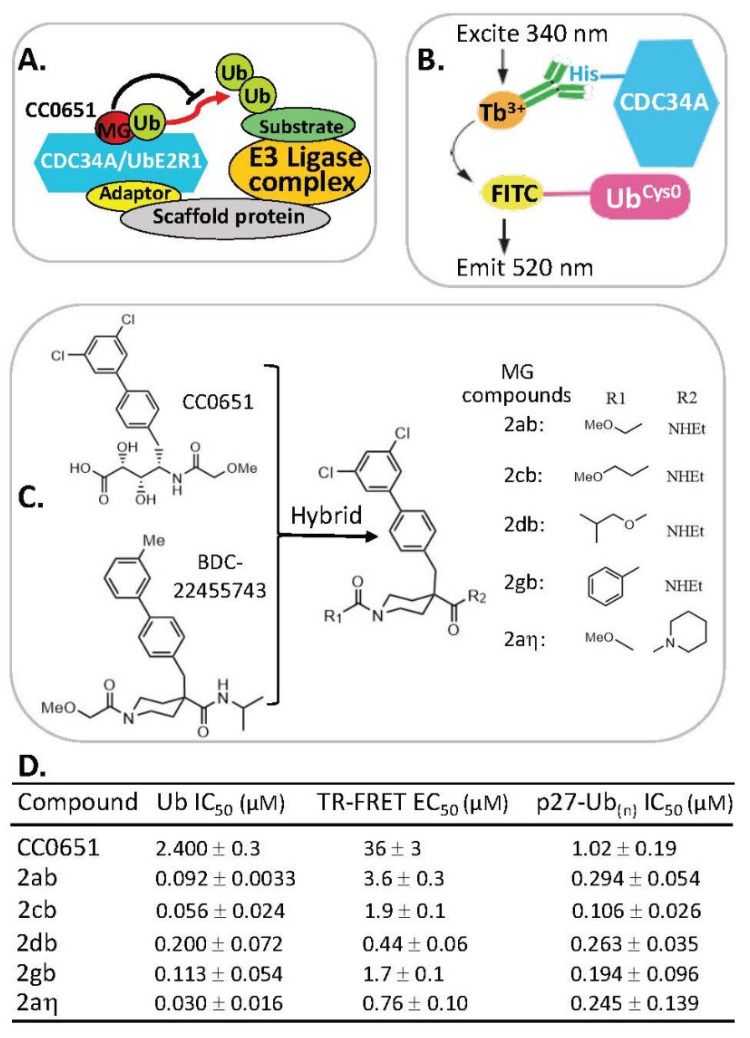
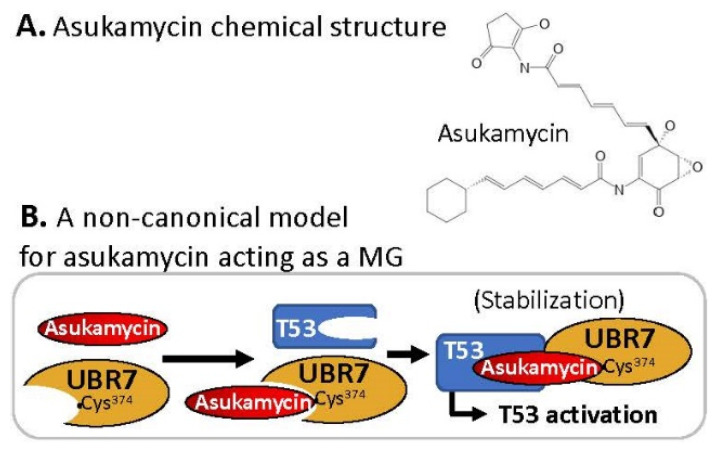
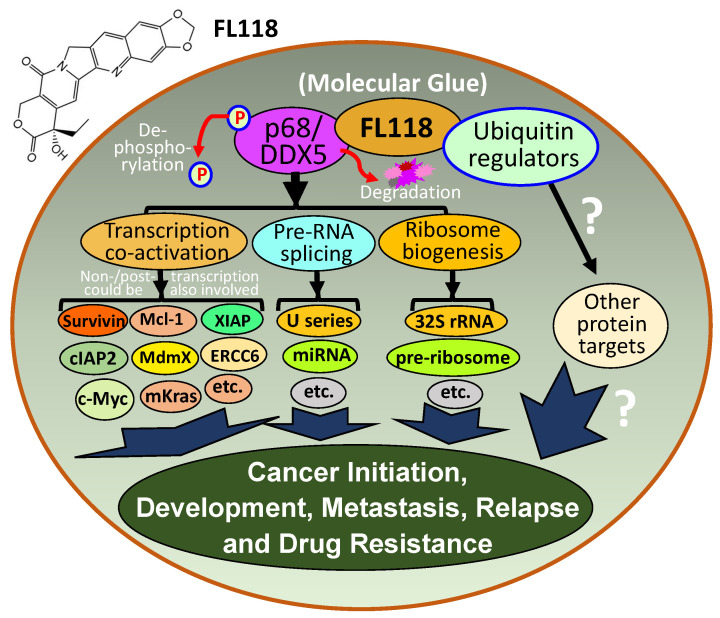
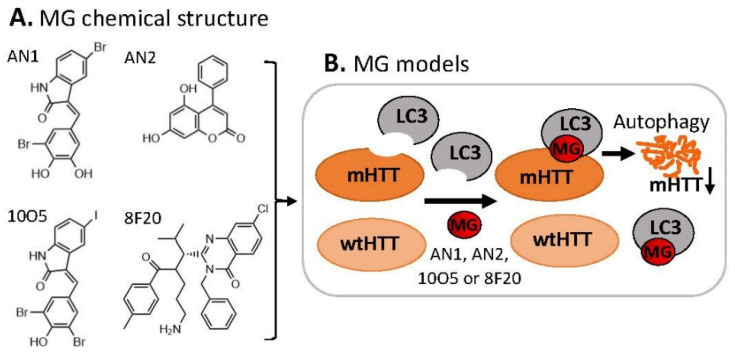
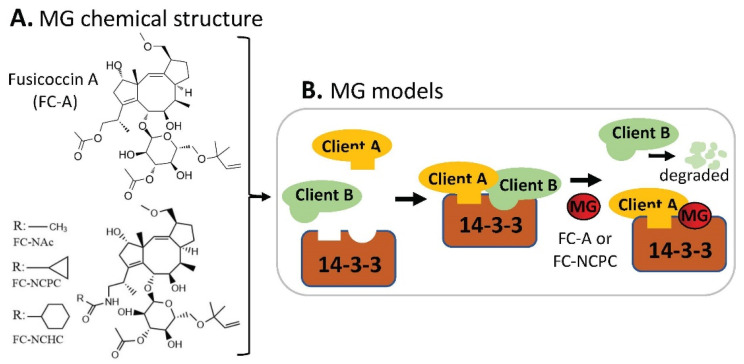
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
