

Glucose-6-Phosphate Dehydrogenase Deficiency and Neonatal Hyperbilirubinemia: Insights on Pathophysiology, Diagnosis, and Gene Variants in Disease Heterogeneity
- PMID: 35685917
- PMCID: PMC9170901
- DOI: 10.3389/fped.2022.875877
Glucose-6-Phosphate Dehydrogenase Deficiency and Neonatal Hyperbilirubinemia: Insights on Pathophysiology, Diagnosis, and Gene Variants in Disease Heterogeneity
Abstract
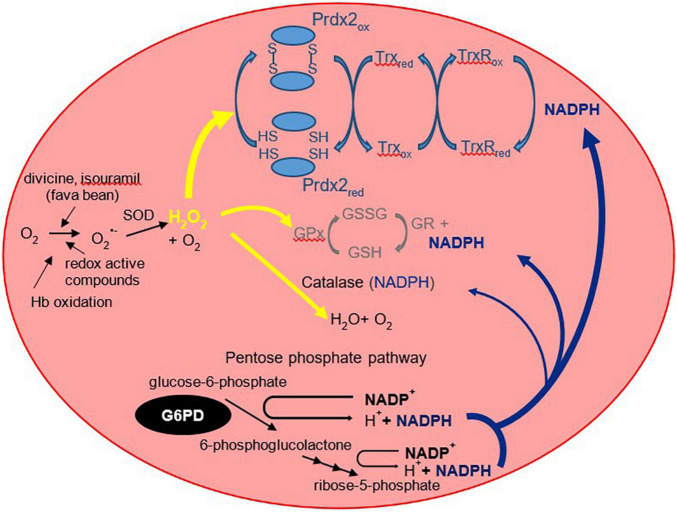
Glucose-6-phosphate dehydrogenase (G6PD) deficiency is a prevalent condition worldwide and is caused by loss-of-function mutations in the G6PD gene. Individuals with deficiency are more susceptible to oxidative stress which leads to the classical, acute hemolytic anemia (favism). However, G6PD deficiency in newborn infants presents with an increased risk of hyperbilirubinemia, that may rapidly escalate to result in bilirubin induced neurologic dysfunction (BIND). Often with no overt signs of hemolysis, G6PD deficiency in the neonatal period appears to be different in the pathophysiology from favism. This review discusses and compares the mechanistic pathways involved in these two clinical presentations of this enzyme disorder. In contrast to the membrane disruption of red blood cells and Heinz bodies formation in favism, G6PD deficiency causing jaundice is perhaps attributed to the disruption of oxidant-antioxidant balance, impaired recycling of peroxiredoxin 2, thus affecting bilirubin clearance. Screening for G6PD deficiency and close monitoring of affected infants are important aspects in neonatal care to prevent kernicterus, a permanent and devastating neurological damage. WHO recommends screening for G6PD activity of all infants in countries with high prevalence of this deficiency. The traditional fluorescent spot test as a screening tool, although low in cost, misses a significant proportion of cases with moderate deficiency or the partially deficient, heterozygote females. Some newer and emerging laboratory tests and diagnostic methods will be discussed while developments in genomics and proteomics contribute to increasing studies that spatially profile genetic mutations within the protein structure that could predict their functional and structural effects. In this review, several known variants of G6PD are highlighted based on the location of the mutation and amino acid replacement. These could provide insights on why some variants may cause a higher degree of phenotypic severity compared to others. Further studies are needed to elucidate the predisposition of some variants toward certain clinical manifestations, particularly neonatal hyperbilirubinemia, and how some variants increase in severity when co-inherited with other blood- or bilirubin-related genetic disorders.
Keywords: G6PD; Heinz bodies; bilirubin; favism; hemolytic anemia; molecular screening; neonatal jaundice; peroxiredoxin 2.
Copyright © 2022 Lee, Ithnin, Azma, Othman, Salvador and Cheah.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Scopes DA, Bautista JM, Naylor CE, Adams MJ, Mason PJ. Amino acid substitutions at the dimer interface of human glucose-6-phosphate dehydrogenase that increase thermostability and reduce the stabilising effect of NADP. Eur J Biochem. (1998) 251:382–8. 10.1046/j.1432-1327.1998.2510382.x - DOI - PubMed
-
- Ainoon O, Alawiyah A, Yu YH, Cheong SK, Hamidah NH, Boo NY, et al. Semiquantitative screening test for G6PD deficiency detects severe deficiency but misses a substantial proportion of partially-deficient females. Southeast Asian J Trop Med Public Health. (2003) 34:405–14. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous