Assembly of complete diploid-phased chromosomes from draft genome sequences
- PMID: 35686922
- PMCID: PMC9339290
- DOI: 10.1093/g3journal/jkac143
Assembly of complete diploid-phased chromosomes from draft genome sequences
Abstract
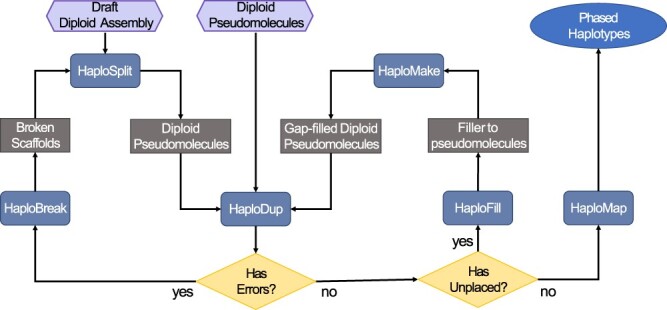
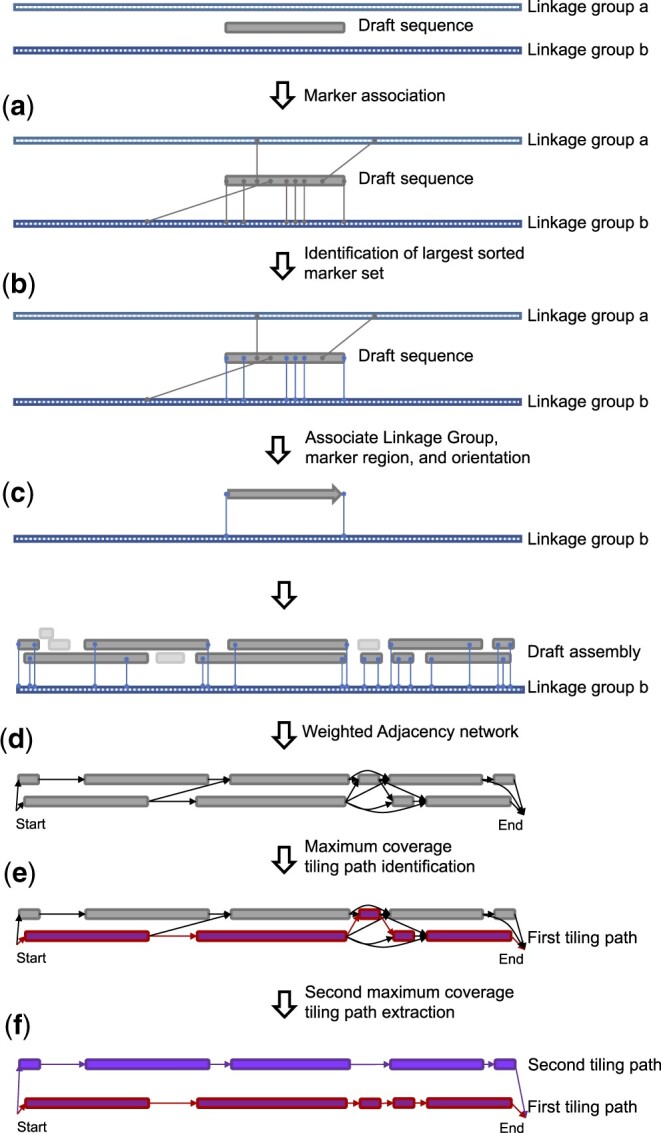
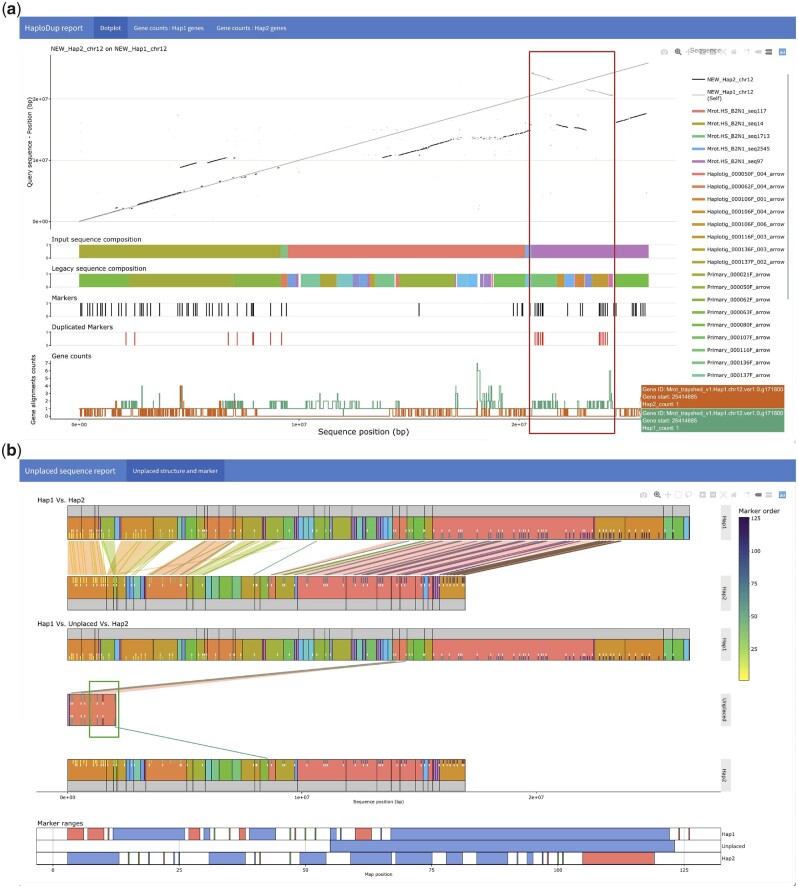
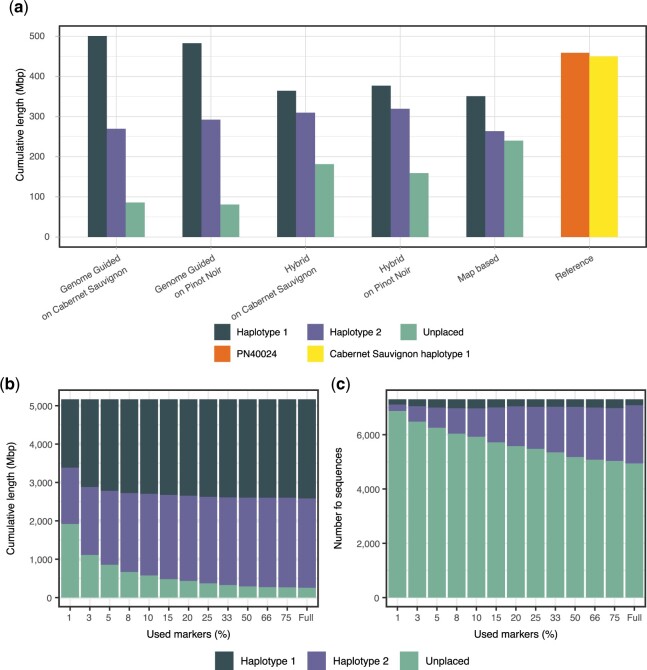
De novo genome assembly is essential for genomic research. High-quality genomes assembled into phased pseudomolecules are challenging to produce and often contain assembly errors because of repeats, heterozygosity, or the chosen assembly strategy. Although algorithms that produce partially phased assemblies exist, haploid draft assemblies that may lack biological information remain favored because they are easier to generate and use. We developed HaploSync, a suite of tools that produces fully phased, chromosome-scale diploid genome assemblies, and performs extensive quality control to limit assembly artifacts. HaploSync scaffolds sequences from a draft diploid assembly into phased pseudomolecules guided by a genetic map and/or the genome of a closely related species. HaploSync generates a report that visualizes the relationships between current and legacy sequences, for both haplotypes, and displays their gene and marker content. This quality control helps the user identify misassemblies and guides Haplosync's correction of scaffolding errors. Finally, HaploSync fills assembly gaps with unplaced sequences and resolves collapsed homozygous regions. In a series of plant, fungal, and animal kingdom case studies, we demonstrate that HaploSync efficiently increases the assembly contiguity of phased chromosomes, improves completeness by filling gaps, corrects scaffolding, and correctly phases highly heterozygous, complex regions.
Keywords: assembly error correction; chromosome anchoring; diploid genomes; haplotype phasing; hybrid genome assembly.
© The Author(s) 2022. Published by Oxford University Press on behalf of Genetics Society of America.
Figures