

Recent advances in isobaric labeling and applications in quantitative proteomics
- PMID: 35687565
- PMCID: PMC9787039
- DOI: 10.1002/pmic.202100256
Recent advances in isobaric labeling and applications in quantitative proteomics
Abstract
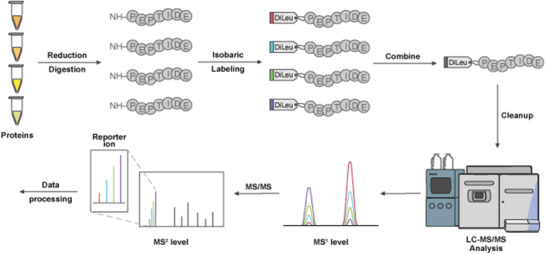
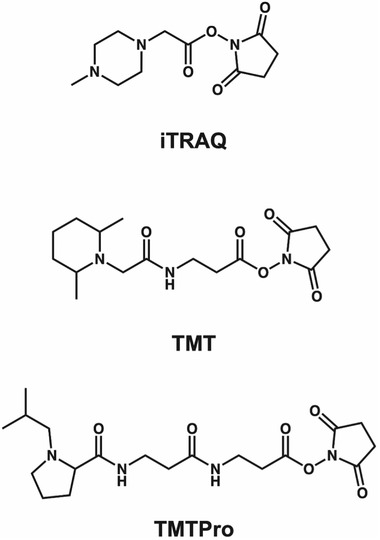
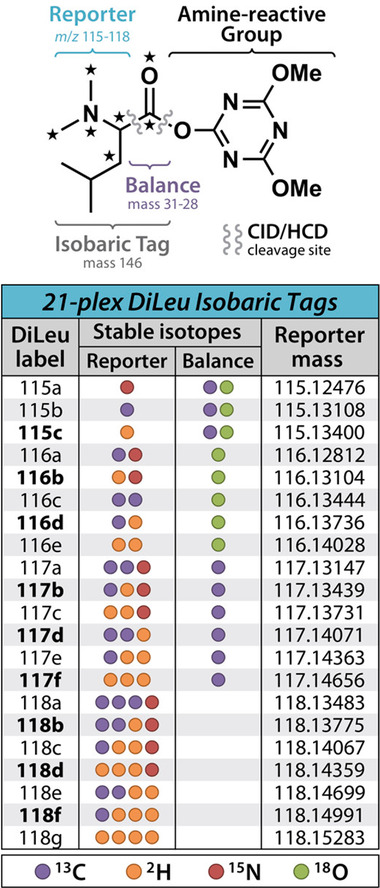
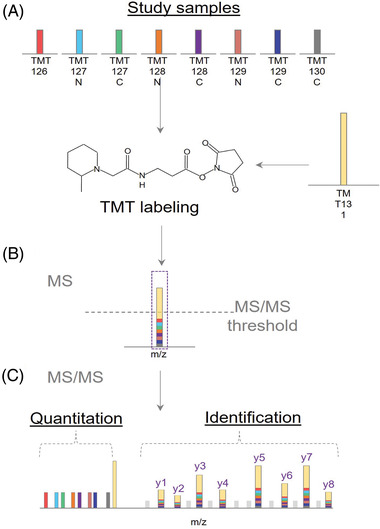
Mass spectrometry (MS) has emerged at the forefront of quantitative proteomic techniques. Liquid chromatography-mass spectrometry (LC-MS) can be used to determine abundances of proteins and peptides in complex biological samples. Several methods have been developed and adapted for accurate quantification based on chemical isotopic labeling. Among various chemical isotopic labeling techniques, isobaric tagging approaches rely on the analysis of peptides from MS2-based quantification rather than MS1-based quantification. In this review, we will provide an overview of several isobaric tags along with some recent developments including complementary ion tags, improvements in sensitive quantitation of analytes with lower abundance, strategies to increase multiplexing capabilities, and targeted analysis strategies. We will also discuss limitations of isobaric tags and approaches to alleviate these restrictions through bioinformatic tools and data acquisition methods. This review will highlight several applications of isobaric tags, including biomarker discovery and validation, thermal proteome profiling, cross-linking for structural investigations, single-cell analysis, top-down proteomics, along with applications to different molecules including neuropeptides, glycans, metabolites, and lipids, while providing considerations and evaluations to each application.
Keywords: isobaric tags; isotopic labeling; mass spectrometry; protein quantitation; quantitative proteomics; systems biology.
© 2022 The Authors. Proteomics published by Wiley-VCH GmbH.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
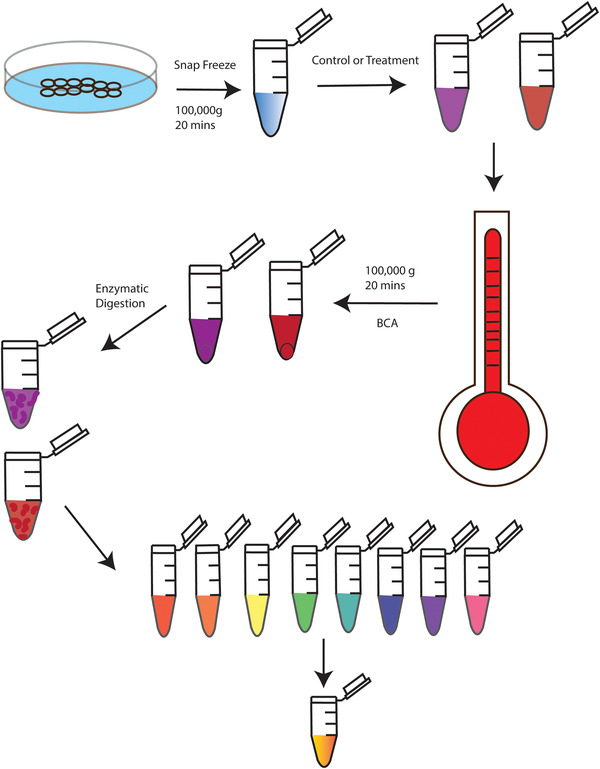
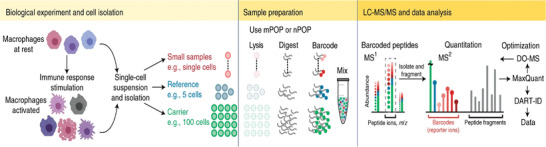
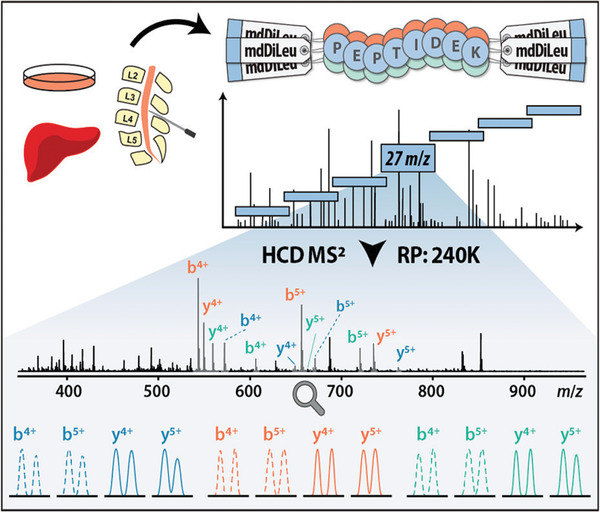
References
-
- Asara, J. M. , Christofk, H. R. , Freimark, L. M. , & Cantley, L. C. (2008). A label‐free quantification method by MS/MS TIC compared to SILAC and spectral counting in a proteomics screen. Proteomics, 8, 994–999. - PubMed
-
- Bondarenko, P. V. , Chelius, D. , & Shaler, T. A. (2002). Identification and relative quantitation of protein mixtures by enzymatic digestion followed by capillary reversed‐phase liquid chromatography−tandem mass spectrometry. Analytical Chemistry, 74(18), 4741–4749. - PubMed
-
- Gygi, S. P. , Rist, B. , Gerber, S. A. , Turecek, F. , Gelb, M. H. , & Aebersold, R. (1999). Quantitative analysis of complex protein mixtures using isotope‐coded affinity tags, Nature Biotechnology, 17, 994–999. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
