

Mapping information-rich genotype-phenotype landscapes with genome-scale Perturb-seq
- PMID: 35688146
- PMCID: PMC9380471
- DOI: 10.1016/j.cell.2022.05.013
Mapping information-rich genotype-phenotype landscapes with genome-scale Perturb-seq
Abstract
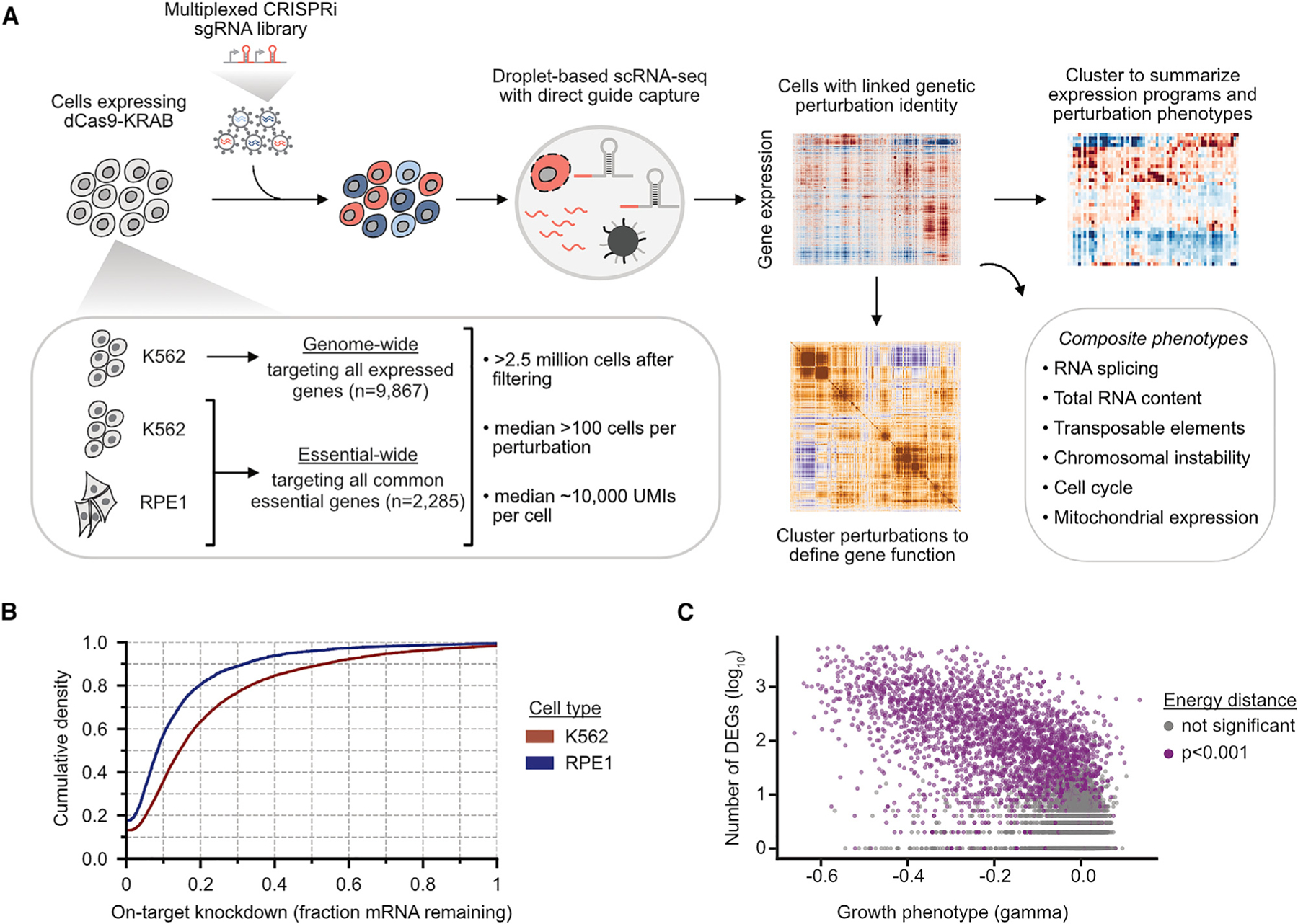
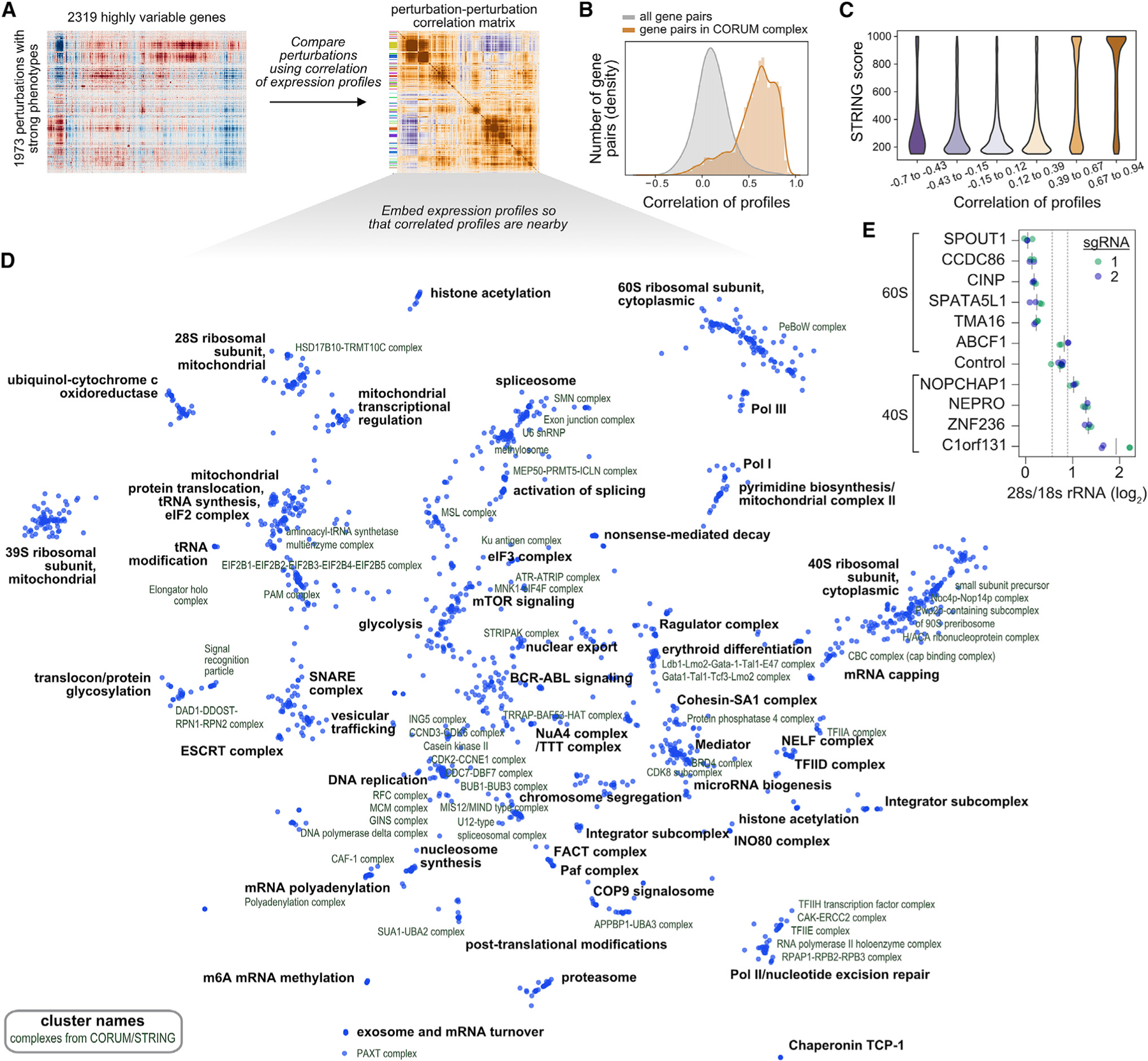
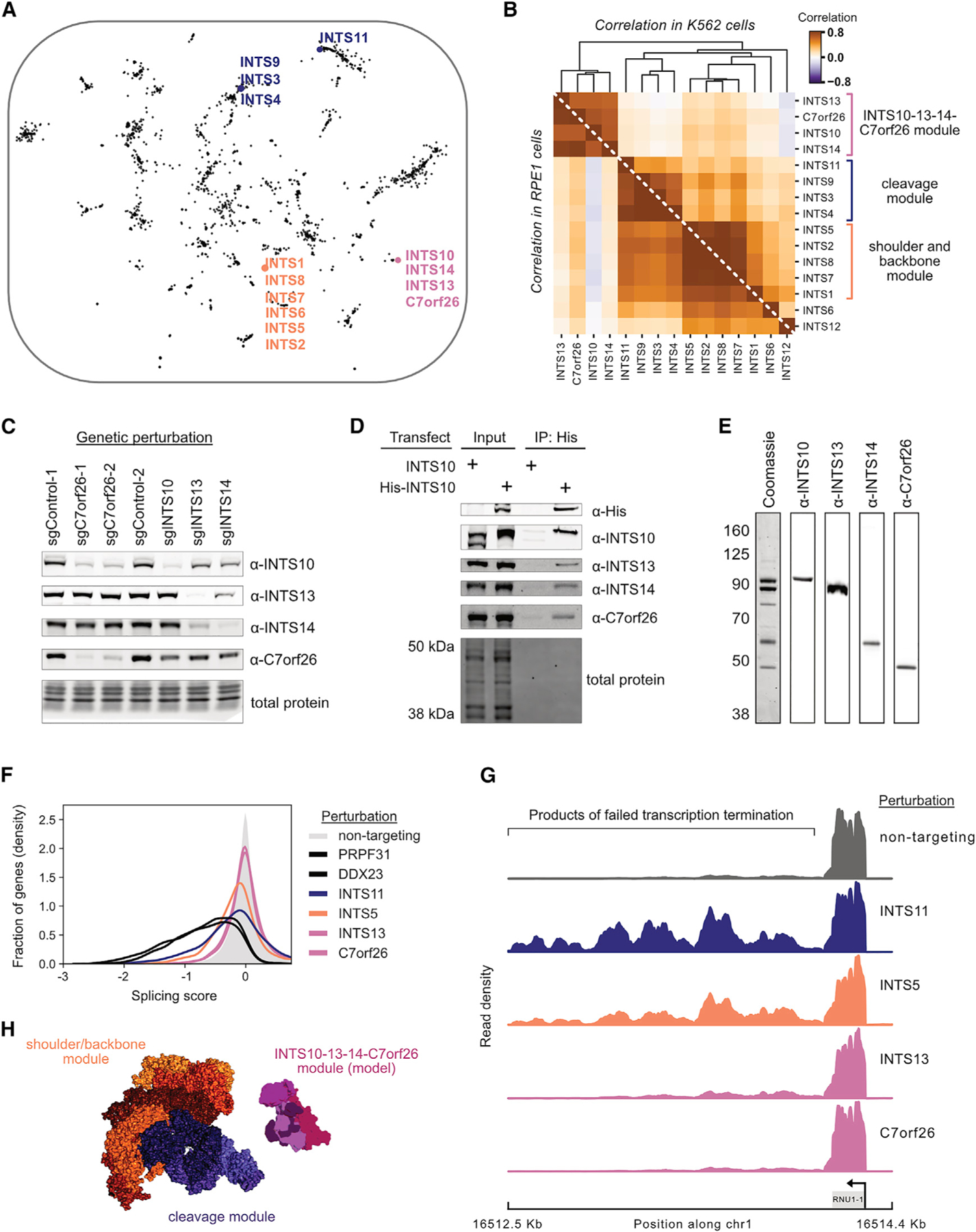
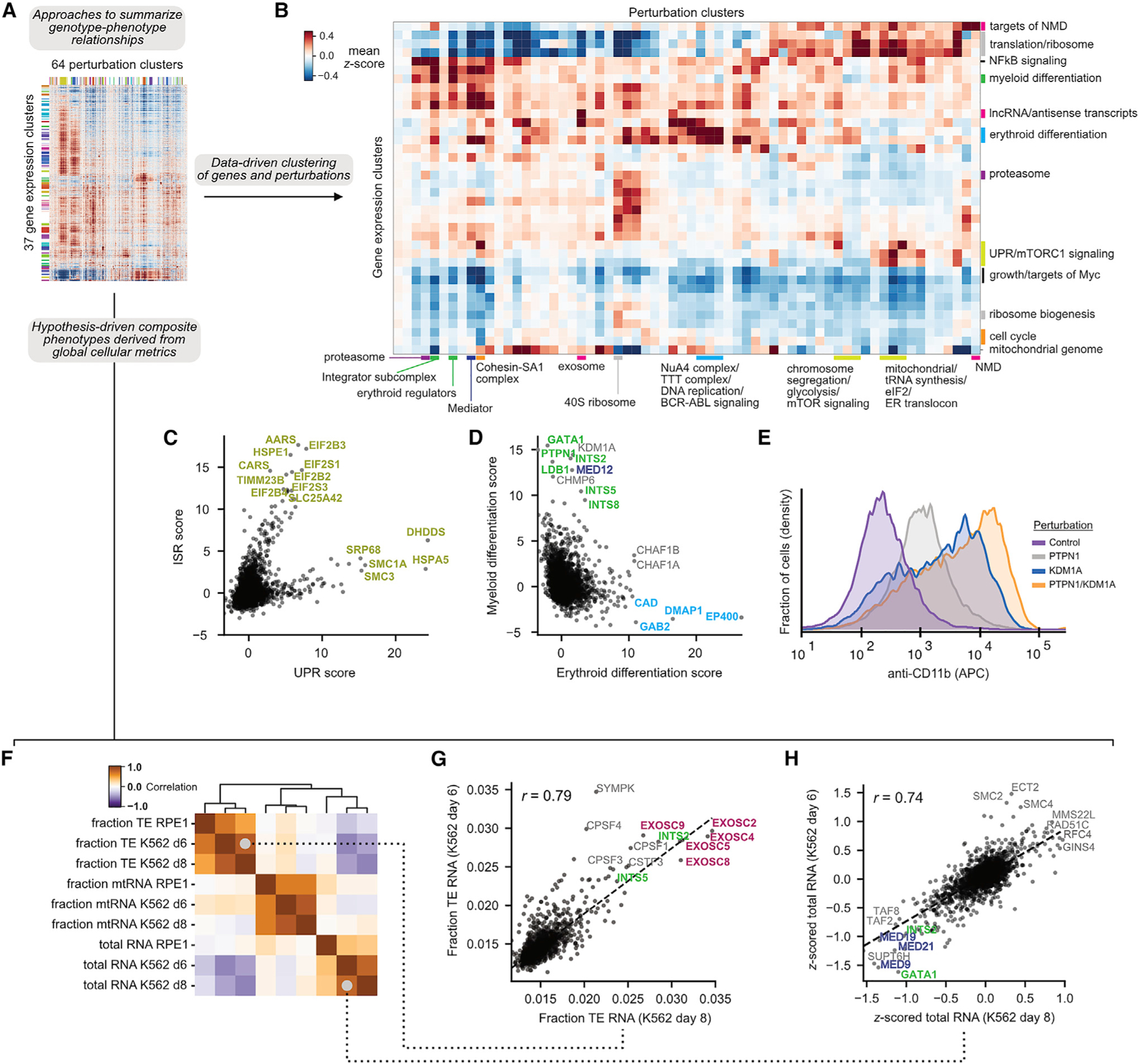
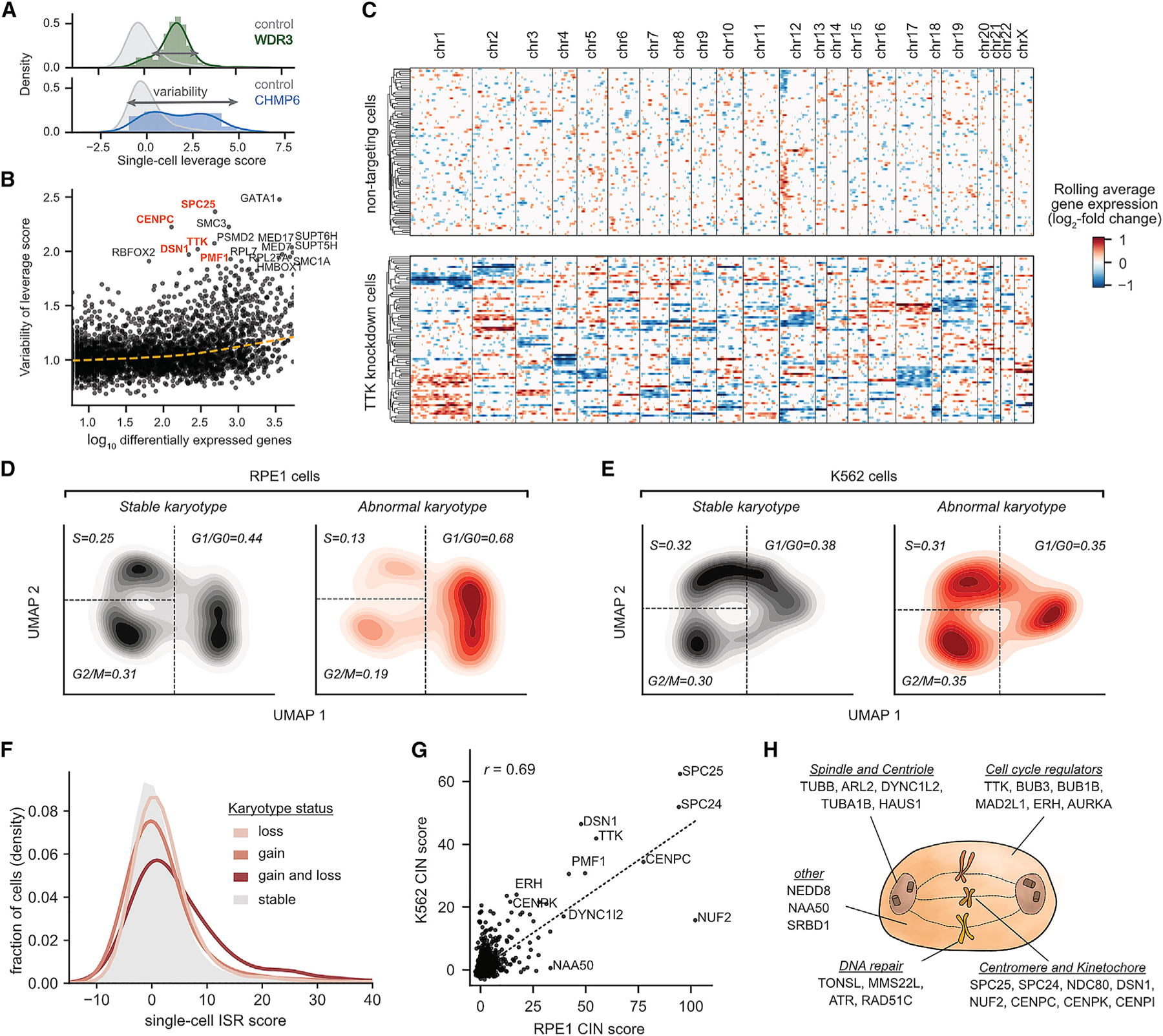
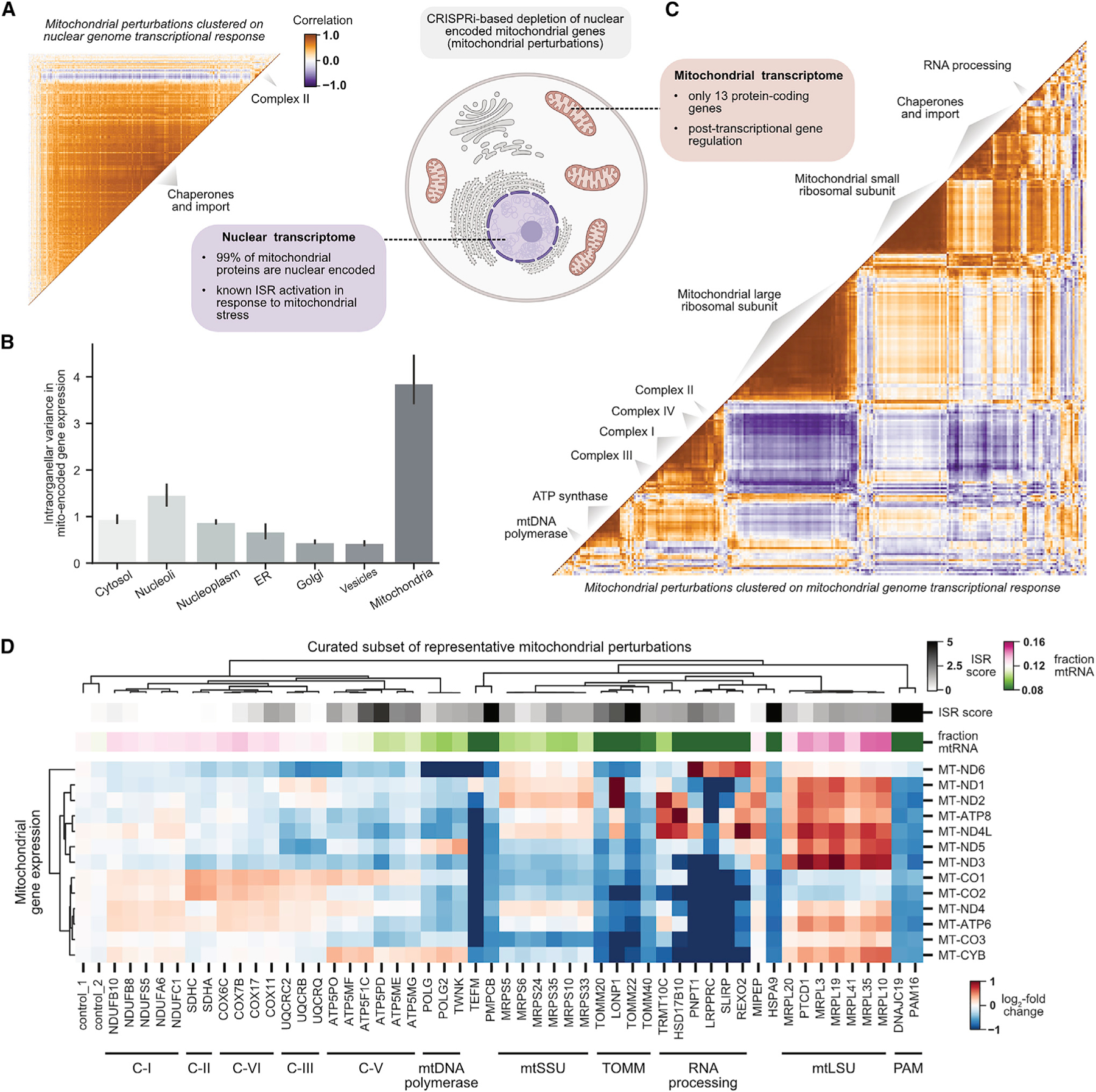
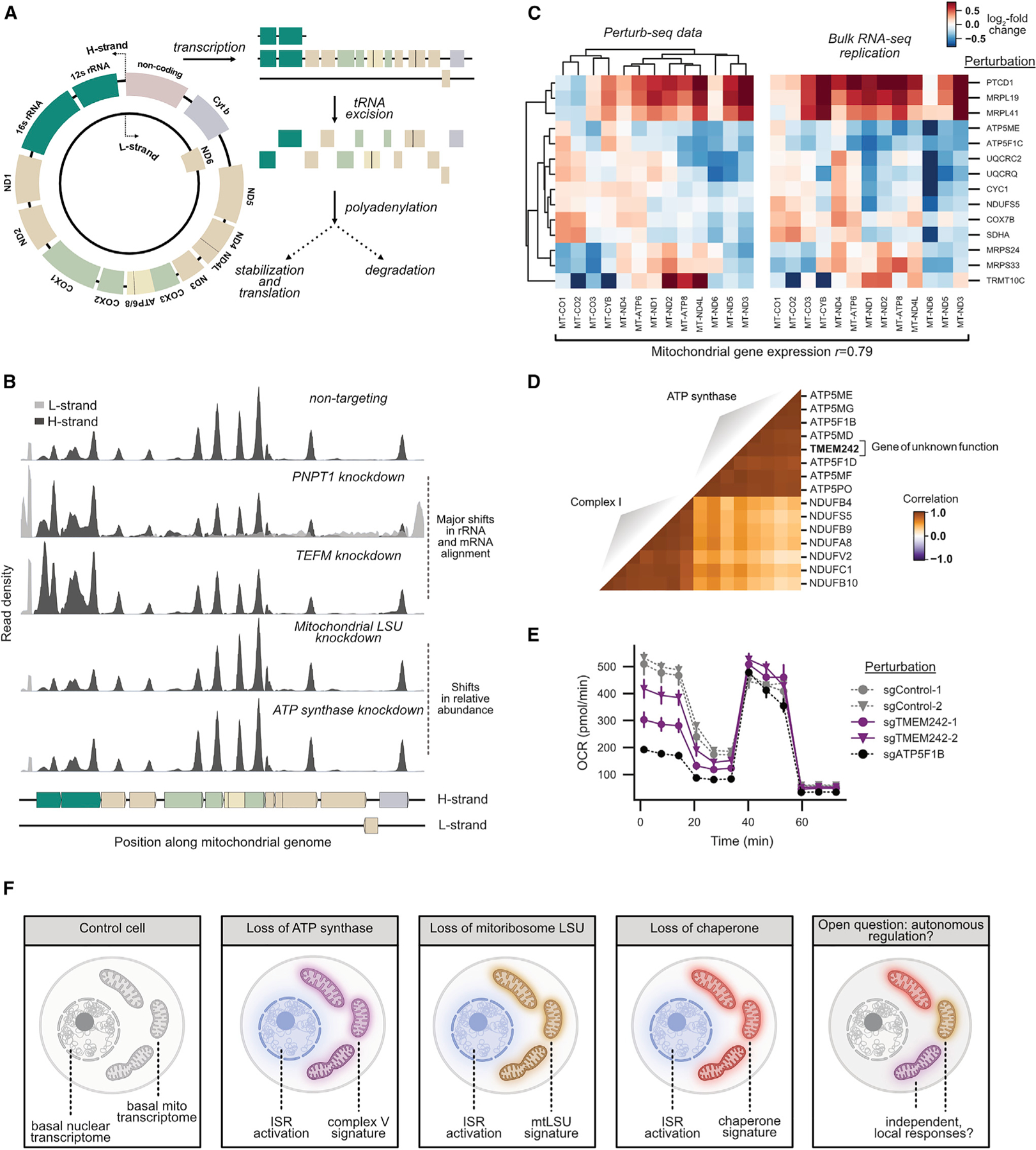
A central goal of genetics is to define the relationships between genotypes and phenotypes. High-content phenotypic screens such as Perturb-seq (CRISPR-based screens with single-cell RNA-sequencing readouts) enable massively parallel functional genomic mapping but, to date, have been used at limited scales. Here, we perform genome-scale Perturb-seq targeting all expressed genes with CRISPR interference (CRISPRi) across >2.5 million human cells. We use transcriptional phenotypes to predict the function of poorly characterized genes, uncovering new regulators of ribosome biogenesis (including CCDC86, ZNF236, and SPATA5L1), transcription (C7orf26), and mitochondrial respiration (TMEM242). In addition to assigning gene function, single-cell transcriptional phenotypes allow for in-depth dissection of complex cellular phenomena-from RNA processing to differentiation. We leverage this ability to systematically identify genetic drivers and consequences of aneuploidy and to discover an unanticipated layer of stress-specific regulation of the mitochondrial genome. Our information-rich genotype-phenotype map reveals a multidimensional portrait of gene and cellular function.
Keywords: CRISPR; Integrator complex; Perturb-seq; cell biology; chromosomal instability; genetic screens; genotype-phenotype map; mitochondrial genome stress response; single-cell RNA sequencing.
Copyright © 2022 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests J.M.R. consults for Maze Therapeutics and is a consultant for and equity holder in Waypoint Bio. R.A.S. consults for Maze Therapeutics. K.A. is a consultant for Syros Pharmaceuticals, is on the SAB of CAMP4 Therapeutics, and received research funding from Novartis not related to this work. G.L.-Y., N.I., F.O., and D.L. are employees and shareholders of Ultima Genomics. M.J. consults for Maze Therapeutics and Gate Bioscience. T.M.N. consults for Maze Therapeutics. J.S.W. declares outside interest in 5AM Ventures, Amgen, Chroma Medicine, KSQ Therapeutics, Maze Therapeutics, Tenaya Therapeutics, Tessera Therapeutics, and Third Rock Ventures. The Regents of the University of California with R.A.S., T.M.N., M.J., and J.S.W. as inventors have filed patent applications related to CRISPRi/a screening and Perturb-seq.
Figures
Comment in
-
Genome-scale single-cell CRISPR screens.Nat Rev Genet. 2022 Aug;23(8):459. doi: 10.1038/s41576-022-00517-1. Nat Rev Genet. 2022. PMID: 35760907 No abstract available.
References
-
- Adamson B, Norman TM, Jost M, Cho MY, Nuñez JK, Chen Y, Villalta JE, Gilbert LA, Horlbeck MA, Hein MY, et al. (2016). A multiplexed single-cell CRISPR screening platform enables systematic dissection of the unfolded protein response. Cell 167, 1867–1882.e21. 10.1016/j.cell.2016.11.048. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
