

Factors shaping the abundance and diversity of the gut archaeome across the animal kingdom
- PMID: 35688919
- PMCID: PMC9187648
- DOI: 10.1038/s41467-022-31038-4
Factors shaping the abundance and diversity of the gut archaeome across the animal kingdom
Abstract
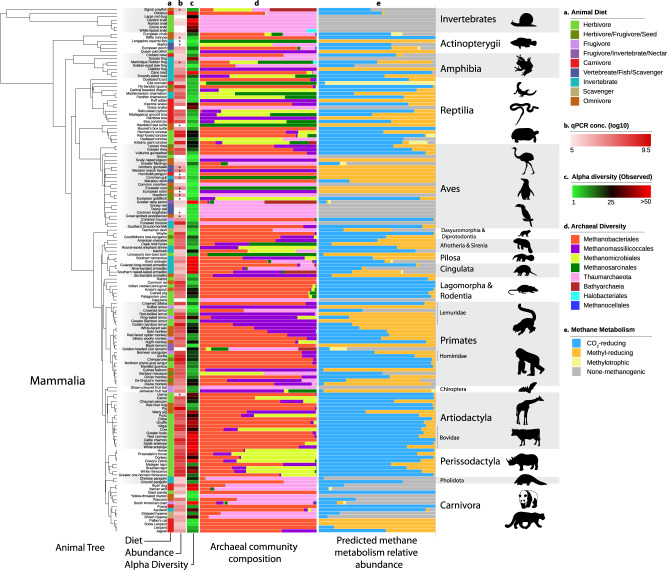
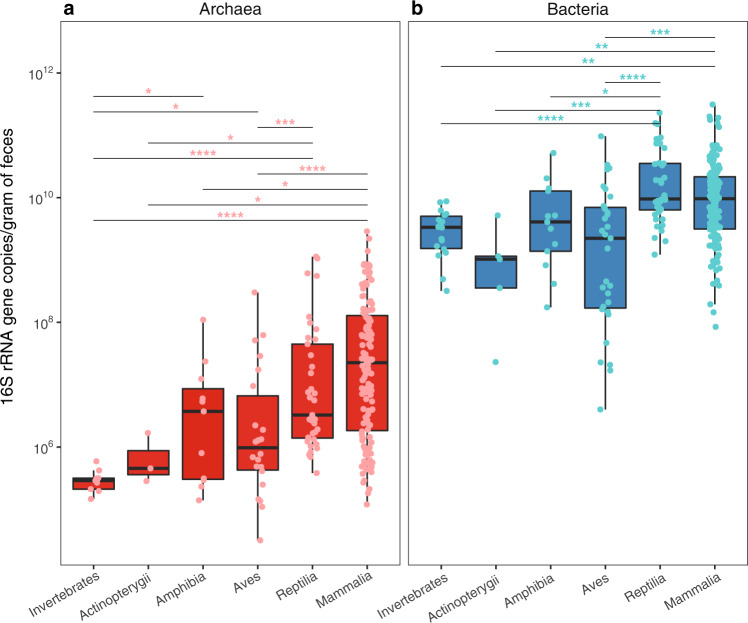
Archaea are common constituents of the gut microbiome of humans, ruminants, and termites but little is known about their diversity and abundance in other animals. Here, we analyse sequencing and quantification data of archaeal and bacterial 16S rRNA genes from 250 species of animals covering a large taxonomic spectrum. We detect the presence of archaea in 175 animal species belonging to invertebrates, fish, amphibians, birds, reptiles and mammals. We identify five dominant gut lineages, corresponding to Methanobrevibacter, Methanosphaera, Methanocorpusculum, Methanimicrococcus and "Ca. Methanomethylophilaceae". Some archaeal clades, notably within Methanobrevibacter, are associated to certain hosts, suggesting specific adaptations. The non-methanogenic lineage Nitrososphaeraceae (Thaumarchaeota) is frequently present in animal samples, although at low abundance, but may have also adapted to the gut environment. Host phylogeny, diet type, fibre content, and intestinal tract physiology are major drivers of the diversity and abundance of the archaeome in mammals. The overall abundance of archaea is more influenced by these factors than that of bacteria. Methanogens reducing methyl-compounds with H2 can represent an important fraction of the overall methanogens in many animals. Together with CO2-reducing methanogens, they are influenced by diet and composition of gut bacteria. Our results provide key elements toward our understanding of the ecology of archaea in the gut, an emerging and important field of investigation.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures





References
-
- Kapitan, M., Niemiec, M. J., Steimle, A., FrickIlse, J. S. & Jacobsen, D. Fungi as Part of the Microbiota and Interactions with Intestinal Bacteria. In Fungal Physiology and Immunopathogenesis422 (Springer, Cham, 2018). - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
