

Oriented internal electrostatic fields: an emerging design element in coordination chemistry and catalysis
- PMID: 35694353
- PMCID: PMC9116365
- DOI: 10.1039/d2sc01715f
Oriented internal electrostatic fields: an emerging design element in coordination chemistry and catalysis
Abstract
The power of oriented electrostatic fields (ESFs) to influence chemical bonding and reactivity is a phenomenon of rapidly growing interest. The presence of strong ESFs has recently been implicated as one of the most significant contributors to the activity of select enzymes, wherein alignment of a substrate's changing dipole moment with a strong, local electrostatic field has been shown to be responsible for the majority of the enzymatic rate enhancement. Outside of enzymology, researchers have studied the impacts of "internal" electrostatic fields via the addition of ionic salts to reactions and the incorporation of charged functional groups into organic molecules (both experimentally and computationally), and "externally" via the implementation of bulk fields between electrode plates. Incorporation of charged moieties into homogeneous inorganic complexes to generate internal ESFs represents an area of high potential for novel catalyst design. This field has only begun to materialize within the past 10 years but could be an area of significant impact moving forward, since it provides a means for tuning the properties of molecular complexes via a method that is orthogonal to traditional strategies, thereby providing possibilities for improved catalytic conditions and novel reactivity. In this perspective, we highlight recent developments in this area and offer insights, obtained from our own research, on the challenges and future directions of this emerging field of research.
This journal is © The Royal Society of Chemistry.
Conflict of interest statement
There are no conflicts to declare.
Figures







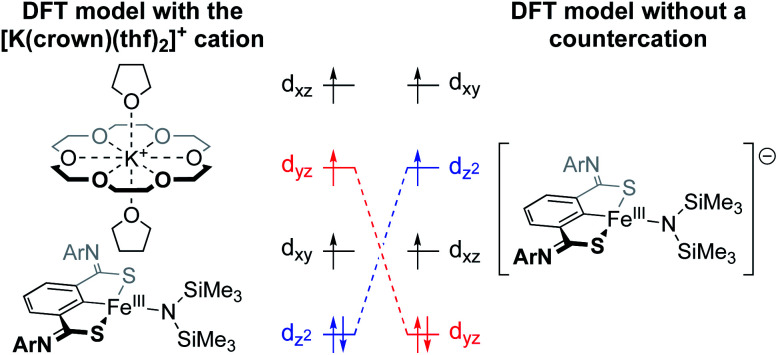









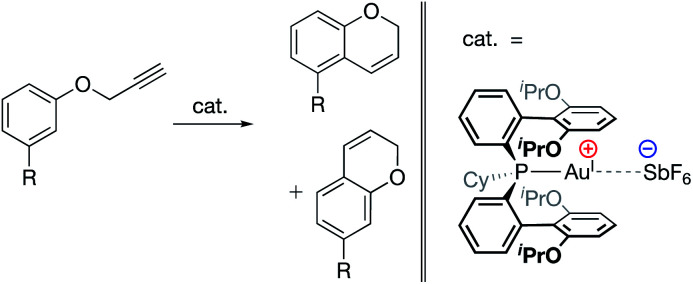





References
-
- Che F. Gray J. T. Ha S. Kruse N. Scott S. L. McEwen J.-S. ACS Catal. 2018;9:5153–5174. doi: 10.1021/acscatal.7b02899. - DOI
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous